|
2013年7月26日,美国食品及药物管理局(FDA)批准新型SNRI类药物Fetzima(levomilnacipran,左旋米那普仑)用于治疗成人重型抑郁障碍,这也是经FDA批准在美国上市的第四种5-羟色胺及去甲肾上腺素再摄取抑制剂。
HIGHLIGHTS OF PRESCRIBING INFORMATION
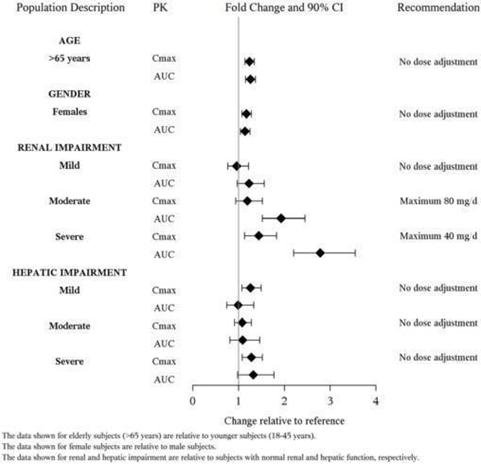
FETZIMA is a serotonin and norepinephrine reuptake inhibitor (SNRI) indicated for the treatment of Major Depressive Disorder (MDD) (1). Limitation of Use: FETZIMA is not approved for the management of fibromyalgia. The efficacy and safety of FETZIMA for the management of fibromyalgia have not been established (1). DOSAGE AND ADMINISTRATION Recommended dose: 40 mg to 120 mg once daily with or without food (2.1). Initiate dose at 20 mg once daily for 2 days and then increase to 40 mg once daily (2.1). Based on efficacy and tolerability, increase dose in increments of 40 mg at intervals of 2 or more days (2.1). The maximum recommended dose is 120 mg once daily (2.1). Take capsules whole; do not open, chew or crush (2.1) Renal Impairment: Do not exceed 80 mg once daily for moderate impairment. Do not exceed 40 mg once daily for severe renal impairment (2.3). Discontinuation: Reduce dose gradually whenever possible (2.4) DOSAGE FORMS AND STRENGTHS Extended-release capsules: 20 mg, 40 mg, 80 mg and 120 mg (3). CONTRAINDICATIONS Hypersensitivity to levomilnacipran, milnacipran HCl, or any excipient in the FETZIMA formulation (4). Serotonin Syndrome and MAOIs: Do not use MAOIs intended to treat psychiatric disorders with FETZIMA or within 7 days of stopping treatment with FETZIMA. Do not use FETZIMA within 14 days of stopping an MAOI intended to treat psychiatric disorders. In addition, do not start FETZIMA in a patient who is being treated with linezolid or intravenous methylene blue (4). WARNINGS AND PRECAUTIONS Suicidal Thoughts and Behaviors in Children, Adolescents, and Young Adults: Monitor patients for clinical worsening and suicidal thinking or behavior (5.1). Serotonin Syndrome: Serotonin syndrome has been reported with SSRIs and SNRIs, both when taken alone, but especially when co-administered with other serotonergic agents (including triptans, tricyclics, fentanyl, lithium, tramadol, tryptophan, buspirone and St. John's Wort). If such symptoms occur, discontinue FETZIMA and initiate supportive treatment. If concomitant use of FETZIMA with other serotonergic drugs is clinically warranted, patients should be made aware of a potential increased risk for serotonin syndrome, particularly during treatment initiation and dose increases (5.2). Elevated Blood Pressure and Heart Rate: Measure heart rate and blood pressure prior to initiating treatment and periodically throughout treatment. Control pre-existing hypertension before initiating therapy with FETZIMA (5.3, 5.4). Abnormal Bleeding: Treatment can increase the risk of bleeding. Caution patients about the risk of bleeding associated with the use of NSAIDs, aspirin, or other drugs that affect coagulation (5.5). Angle Closure Glaucoma: Angle closure glaucoma has occurred in patients with untreated anatomically narrow angles treated with antidepressants (5.6). Urinary Hesitation or Retention: Can occur. If such symptoms occur, discontinue FETZIMA or consider other appropriate medical intervention (5.7). Activation of Mania/Hypomania: Screen patients for bipolar disorder, Caution patients about risk of activation of mania/hypomania (5.8). Seizures: Can occur. Use with caution in patients with a seizure disorder (5.9). Discontinuation Syndrome: Taper dose when possible and monitor for discontinuation symptoms (5.10). Hyponatremia: Can occur in association with SIADH (5.11). ADVERSE REACTIONS The most common adverse reactions (incidence ≥ 5% and at least twice the rate of placebo) are: nausea, constipation, hyperhidrosis, heart rate increase, erectile dysfunction, tachycardia, vomiting, and palpitations (6.1). To report SUSPECTED ADVERSE REACTIONS, contact Forest Pharmaceuticals, Inc. at 1-800-678-1605 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS Strong CYP3A4 inhibitors such as ketoconazole: Do not exceed 80 mg once daily (7). USE IN SPECIFIC POPULATIONS Pregnancy: Based on animal data, may cause fetal harm (8.1). See 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 1/2016 FULL PRESCRIBING INFORMATION: CONTENTS* 1 INDICATIONS AND USAGE FETZIMA, a serotonin and norepinephrine reuptake inhibitor (SNRI) is indicated for the treatment of major depressive disorder (MDD). The efficacy of FETZIMA was established in three 8-week, randomized, double-blind, placebo-controlled studies in adult patients with a diagnosis of MDD [see Clinical Studies (14)]. Limitation of Use: FETZIMA is not approved for the management of fibromyalgia. The efficacy and safety of FETZIMA for the management of fibromyalgia have not been established. 2 DOSAGE AND ADMINISTRATION 2.1 General Instruction for Use The recommended dose range for FETZIMA is 40 mg to 120 mg once daily, with or without food. FETZIMA should be initiated at 20 mg once daily for 2 days and then increased to 40 mg once daily. Based on efficacy and tolerability, FETZIMA may then be increased in increments of 40 mg at intervals of 2 or more days. The maximum recommended dose is 120 mg once daily. FETZIMA should be taken at approximately the same time each day. FETZIMA should be swallowed whole. Do not open, chew or crush the capsule. 2.2 Maintenance/Continuation/Extended Treatment It is generally agreed that acute episodes of major depressive disorder require several months or longer of sustained pharmacologic therapy. Patients should be reassessed periodically to determine the need for maintenance treatment and the appropriate dose for treatment. The efficacy of FETZIMA has not been established beyond 8 weeks. 2.3 Special Populations Renal Impairment: Dose adjustment is not recommended in patients with mild renal impairment (creatinine clearance of 60-89 mL/min). For patients with moderate renal impairment (creatinine clearance of 30-59 mL/min), the maintenance dose should not exceed 80 mg once daily. For patients with severe renal impairment (creatinine clearance of 15-29 mL/min), the maintenance dose should not exceed 40 mg once daily. FETZIMA is not recommended for patients with end stage renal disease [see Use in Specific Populations (8.7)]. 2.4 Discontinuing Treatment Discontinuation symptoms have been reported with discontinuation of serotonergic drugs such as FETZIMA. Gradual dose reduction is recommended, instead of abrupt discontinuation, whenever possible. Monitor patients for these symptoms when discontinuing FETZIMA. If intolerable symptoms occur following a dose decrease or upon discontinuation of treatment, consider resuming the previously prescribed dose and decreasing the dose at a more gradual rate [see Warnings and Precautions (5.10)]. 2.5 Switching a Patient To or From a Monoamine Oxidase Inhibitor (MAOI) Intended to Treat Psychiatric Disorders At least 14 days should elapse between discontinuation of an MAOI intended to treat psychiatric disorders and initiation of therapy with FETZIMA. Conversely, at least 7 days should be allowed after stopping FETZIMA before starting an MAOI antidepressant [see Contraindications (4)]. 2.6 Use of FETZIMA with Other MAOIs such as Linezolid or Methylene Blue Do not start FETZIMA in a patient who is being treated with linezolid or intravenous methylene blue because there is an increased risk of serotonin syndrome. In a patient who requires more urgent treatment of a psychiatric condition, other interventions, including hospitalization, should be considered [see Contraindications (4)]. In some cases, a patient already receiving FETZIMA therapy may require urgent treatment with linezolid or intravenous methylene blue. If acceptable alternatives to linezolid or intravenous methylene blue treatment are not available and the potential benefits of linezolid or intravenous methylene blue treatment are judged to outweigh the risks of serotonin syndrome in a particular patient, FETZIMA should be stopped promptly, and linezolid or intravenous methylene blue can be administered. The patient should be monitored for symptoms of serotonin syndrome for 2 weeks or until 24 hours after the last dose of linezolid or intravenous methylene blue, whichever comes first. Therapy with FETZIMA may be resumed 24 hours after the last dose of linezolid or intravenous methylene blue [see Warnings and Precautions (5.2)]. The risk of administering methylene blue by non-intravenous routes (such as oral tablets or by local injection) or in intravenous doses much lower than 1 mg/kg with FETZIMA is unclear. The clinician should, nevertheless, be aware of the possibility of emergent symptoms of serotonin syndrome with such use [see Warnings and Precautions (5.2)]. 2.7 Use of FETZIMA with Strong Inhibitors of Cytochrome P450 (CYP3A4) Enzyme The dose of FETZIMA should not exceed 80 mg once daily when used with strong CYP3A4 inhibitors (e.g. ketoconazole, clarithromycin, ritonavir) [see Drug Interactions (7.4)] 3 DOSAGE FORMS AND STRENGTHS FETZIMA (levomilnacipran) is available as 20 mg, 40 mg, 80 mg and 120 mg extended-release capsules.
Hypersensitivity to levomilnacipran, milnacipran HCl or to any excipient in the formulation. The use of MAOIs intended to treat psychiatric disorders with FETZIMA or within 7 days of stopping treatment with FETZIMA is contraindicated because of an increased risk of serotonin syndrome. The use of FETZIMA within 14 days of stopping an MAOI intended to treat psychiatric disorders is also contraindicated [see Dosage and Administration (2.5) and Warnings and Precautions (5.2)]. Starting FETZIMA in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue is also contraindicated because of an increased risk of serotonin syndrome [see Dosage and Administration (2.6) and Warnings and Precautions (5.2)]. 5 WARNINGS AND PRECAUTIONS 5.1 Suicidal Thoughts and Behaviors in Children, Adolescents, and Young Adults Patients with major depressive disorder (MDD), both adult and pediatric, may experience worsening of their depression and/or the emergence of suicidal ideation and behavior (suicidality) or unusual changes in behavior, whether or not they are taking antidepressant medications, and this risk may persist until significant remission occurs. Suicide is a known risk of depression and certain other psychiatric disorders, and these disorders themselves are the strongest predictors of suicide. There has been a longstanding concern, however, that antidepressants may have a role in inducing worsening of depression and the emergence of suicidality in certain patients during the early phase of treatment. Pooled analyses of short-term placebo-controlled studies of antidepressant drugs (selective serotonin reuptake inhibitors [SSRIs] and others) showed that these drugs increase the risk of suicidal thinking and behavior (suicidality) in children, adolescents, and young adults (ages 18-24) with MDD and other psychiatric disorders. Short-term studies did not show an increase in the risk of suicidality with antidepressants compared to placebo in adults beyond age 24; there was a reduction with antidepressants compared to placebo in adults aged 65 and older. The pooled analyses of placebo-controlled studies in children and adolescents with MDD, obsessive compulsive disorder (OCD), or other psychiatric disorders included a total of 24 short-term studies of 9 antidepressant drugs in over 4400 patients. The pooled analyses of placebo controlled studies in adults with MDD or other psychiatric disorders included a total of 295 short-term studies (median duration of 2 months) of 11 antidepressant drugs in over 77,000 patients. There was considerable variation in risk of suicidality among drugs, but a tendency toward an increase in the younger patients for almost all drugs studied. There were differences in absolute risk of suicidality across the different indications, with the highest incidence in MDD. The risk differences (drug vs. placebo), however, were relatively stable within age strata and across indications. These risk differences (drug-placebo difference in the number of cases of suicidality per 1000 patients treated) are provided in Table 1. Table 1
It is unknown whether the suicidality risk extends to longer-term use, i.e., beyond several months. However, there is substantial evidence from placebo-controlled maintenance studies in adults with depression that the use of antidepressants can delay the recurrence of depression. All patients being treated with antidepressants for any indication should be monitored appropriately and observed closely for clinical worsening, suicidality, and unusual changes in behavior, especially during the initial few months of a course of drug therapy, or at times of dose changes, either increases or decreases. The following symptoms: anxiety, agitation, panic attacks, insomnia, irritability, hostility, aggressiveness, impulsivity, akathisia (psychomotor restlessness), hypomania, and mania, have been reported in adult and pediatric patients being treated with antidepressants for major depressive disorder as well as for other indications, both psychiatric and nonpsychiatric. Although a causal link between the emergence of such symptoms and either the worsening of depression and/or the emergence of suicidal impulses has not been established, there is concern that such symptoms may represent precursors to emerging suicidality. Consideration should be given to changing the therapeutic regimen, including possibly discontinuing the medication, in patients whose depression is persistently worse, or who are experiencing emergent suicidality or symptoms that might be precursors to worsening depression or suicidality, especially if these symptoms are severe, abrupt in onset, or were not part of the patient's presenting symptoms. If the decision has been made to discontinue treatment, medication should be tapered, as rapidly as is feasible, but with recognition that abrupt discontinuation can be associated with certain symptoms [see Dosage and Administration (2.4) and Warnings and Precautions (5.10) for a description of the risks of discontinuation of FETZIMA]. Families and caregivers of patients being treated with antidepressants for major depressive disorder or other indications, both psychiatric and nonpsychiatric, should be alerted about the need to monitor patients for the emergence of agitation, irritability, unusual changes in behavior, and the other symptoms described above, as well as the emergence of suicidality, and to report such symptoms immediately to healthcare providers. Such monitoring should include daily observation by families and caregivers. Prescriptions for FETZIMA should be written for the smallest quantity of capsules consistent with good patient management, in order to reduce the risk of overdose. Screening patients for bipolar disorder A major depressive episode may be the initial presentation of bipolar disorder. It is generally believed (though not established in controlled studies) that treating such an episode with an antidepressant alone may increase the likelihood of precipitation of a mixed/manic episode in patients at risk for bipolar disorder. Whether any of the symptoms described above represent such a conversion is unknown. However, prior to initiating treatment with an antidepressant, patients with depressive symptoms should be adequately screened to determine if they are at risk for bipolar disorder; such screening should include a detailed psychiatric history, including a family history of suicide, bipolar disorder, and depression. It should be noted that FETZIMA is not approved for use in treating bipolar depression. 5.2 Serotonin Syndrome The development of a potentially life-threatening serotonin syndrome has been reported with SNRIs and SSRIs, alone but particularly with concomitant use of other serotonergic drugs (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, and St. John's Wort) and with drugs that impair metabolism of serotonin (in particular, MAOIs, both those intended to treat psychiatric disorders and also others, such as linezolid and intravenous methylene blue). Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Patients should be monitored for the emergence of serotonin syndrome. The concomitant use of FETZIMA with MAOIs intended to treat psychiatric disorders is contraindicated. FETZIMA should also not be started in a patient who is being treated with MAOIs such as linezolid or intravenous methylene blue. All reports with methylene blue that provided information on the route of administration involved intravenous administration in the dose range of 1 mg/kg to 8 mg/kg. No reports involved the administration of methylene blue by other routes (such as oral tablets or local tissue injection) or at lower doses. There may be circumstances when it is necessary to initiate treatment with a MAOI such as linezolid or intravenous methylene blue in a patient taking FETZIMA. FETZIMA should be discontinued before initiating treatment with the MAOI [see Dosage and Administration (2.5, 2.6) and Contraindications (4)]. If concomitant use of FETZIMA with other serotonergic drugs, including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, buspirone, tryptophan, and St. John's Wort is clinically warranted, patients should be made aware of a potential increased risk for serotonin syndrome, particularly during treatment initiation and dose increases. Treatment with FETZIMA and any concomitant serotonergic agents, should be discontinued immediately if the above events occur and supportive symptomatic treatment should be initiated. 5.3 Elevated Blood Pressure SNRIs, including FETZIMA, have been associated with increases in blood pressure. Blood pressure should be measured prior to initiating treatment and periodically throughout FETZIMA treatment. Pre-existing hypertension should be controlled before initiating treatment with FETZIMA. Caution should be exercised in treating patients with pre-existing hypertension, cardiovascular, or cerebrovascular conditions that might be compromised by increases in blood pressure. For patients who experience a sustained increase in blood pressure while receiving FETZIMA, discontinuation or other appropriate medical intervention should be considered. Table 2 shows the mean changes in blood pressure, sustained hypertension, and upward shifts in hypertensive status that were observed in FETZIMA-treated patients in the short-term placebo-controlled studies. Table 2 Blood Pressure Mean Changes, Sustained Hypertension, and Upward Shifts in Hypertensive Status
Pre-hypertension: SBP≥120 mm Hg and ≤139 mm Hg or DBP≥ 80 mm Hg and ≤89 mm Hg Stage I hypertension: SBP≥140 mm Hg and ≤159mm Hg or DBP≥ 90 mm Hg and ≤99 mm Hg Stage II hypertension: SBP≥160 mm Hg or DBP≥100mm Hg In the short-term, placebo-controlled MDD studies, the mean increase from initiation of treatment in systolic BP was 3 mm Hg and diastolic BP was 3.2 mm Hg, as compared to no change in the placebo group. There were no dose-related changes in systolic and diastolic blood pressure observed. In patients exposed to one-year, open-label treatment of FETZIMA (doses range from 40-120 mg once daily), the mean change from initiation of treatment in systolic BP was 3.9 mm Hg and diastolic BP was 3.1 mm Hg. In the short-term, placebo-controlled studies, 11.6 % of patients met orthostatic hypotension criteria (SBP or DBP) in the FETZIMA group compared to 9.7% in the placebo group. Orthostatic reductions of blood pressure ≥ 10 mm Hg in DBP occurred in 5.8%, 6.1% and 9.8% of FETZIMA-treated patients with doses of 40, 80 and 120 mg/day respectively, compared to 6.2% of placebo-treated patients. Concomitant use of FETZIMA with drugs that increase blood pressure and heart rate has not been evaluated and such combinations should be used with caution. Effects of FETZIMA on blood pressure in patients with significant hypertension or cardiac disease have not been systematically evaluated. FETZIMA should be used with caution in these patients. 5.4 Elevated Heart Rate SNRIs including FETZIMA have been associated with increased heart rate. Heart rate should be measured prior to initiating treatment and periodically throughout FETZIMA treatment. Pre-existing tachyarrhythmias and other cardiac disease should be treated before starting therapy with FETZIMA. For patients who experience a sustained increase in heart rate while receiving FETZIMA, discontinuation or other appropriate medical intervention should be considered. In short-term clinical studies, FETZIMA treatment was associated with a mean increase in heart rate of 7.4 beats per minute (bpm) compared to a mean decrease of 0.3 bpm in placebo-treated patients. Heart rate increase in FETZIMA-treated patients receiving doses of 40 mg, 80 mg and 120 mg was 7.2, 7.2, and 9.1 bpm. FETZIMA has not been systematically evaluated in patients with a cardiac rhythm disorder. 5.5 Abnormal Bleeding SSRIs and SNRIs, including FETZIMA, may increase the risk of bleeding events. Concomitant use of aspirin, nonsteroidal anti-inflammatory drugs (NSAIDS), warfarin, and other anticoagulants may add to this risk. Case reports and epidemiological studies (case-control and cohort design) have demonstrated an association between use of drugs that interfere with serotonin reuptake and the occurrence of gastrointestinal bleeding. Bleeding events related to SSRIs and SNRIs have ranged from ecchymosis, hematoma, epistaxis, and petechiae to life-threatening hemorrhages. Patients should be cautioned about the risk of bleeding associated with the concomitant use of FETZIMA and NSAIDs, aspirin, or other drugs that affect coagulation or bleeding. 5.6 Angle Closure Glaucoma The pupillary dilation that occurs following use of many antidepressant drugs including FETZIMA may trigger an angle closure attack in a patient with anatomically narrow angles who does not have a patent iridectomy. 5.7 Urinary Hesitation or Retention The noradrenergic effect of SNRIs including FETZIMA, can affect urethral resistance. In the controlled short-term studies, urinary hesitation occurred in 4%, 5% and 6% of FETZIMA-treated patients receiving doses of 40, 80 and 120 mg, respectively, compared to no patients in the placebo group. Caution is advised in the use of FETZIMA in patients prone to obstructive urinary disorders. If symptoms of urinary hesitation, urinary retention, or dysuria develop during treatment with FETZIMA, consideration should be given to the possibility that they might be drug-related, and discontinuation or other appropriate medical intervention should be considered. 5.8 Activation of Mania/Hypomania Symptoms of mania/hypomania were reported in 0.2% of FETZIMA-treated patients and 0.2% of placebo-treated patients in clinical studies. Activation of mania/hypomania has also been reported in a small proportion of patients with mood disorders who were treated with other antidepressants. As with all antidepressants, use FETZIMA cautiously in patients with a history or family history of bipolar disorder, mania, or hypomania. 5.9 Seizures FETZIMA has not been systematically evaluated in patients with a seizure disorder. Patients with a history of seizures were excluded from clinical studies. FETZIMA should be prescribed with caution in patients with a seizure disorder. One case of seizure has been reported in pre-marketing clinical studies with FETZIMA. 5.10 Discontinuation Syndrome There have been reports of adverse events occurring upon discontinuation of serotonergic antidepressants, particularly when discontinuation is abrupt, including the following: dysphoric mood, irritability, agitation, dizziness, sensory disturbances (e.g., paresthesia, such as electric shock sensations), anxiety, confusion, headache, lethargy, emotional lability, insomnia, hypomania, tinnitus, and seizures. While these events are generally self-limiting, there have been reports of serious discontinuation symptoms. Monitor patients for these symptoms when discontinuing FETZIMA. Reduce the dose gradually whenever possible. If intolerable symptoms occur following a decrease in the dose or upon discontinuation of treatment, consider resuming the previously prescribed dose. Subsequently, the dose may be decreased, but at a more gradual rate [see Dosage and Administration (2.4)]. 5.11 Hyponatremia Although no adverse events of hyponatremia were reported for FETZIMA-treated patients in the clinical studies, hyponatremia has occurred as a result of treatment with SSRIs and SNRIs. In many cases, hyponatremia appears to be the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH). Cases with serum sodium lower than 110 mmol/L have been reported. Elderly patients may be at greater risk of developing hyponatremia with SSRIs and SNRIs. Also, patients taking diuretics or who are otherwise volume depleted can be at greater risk. FETZIMA should be discontinued in patients with symptomatic hyponatremia and appropriate medical intervention should be instituted. Signs and symptoms of hyponatremia include headache, difficulty concentrating, memory impairment, confusion, weakness, and unsteadiness, which can lead to falls. Signs and symptoms associated with more severe and/or acute cases have included hallucination, syncope, seizure, coma, respiratory arrest, and death. 6 ADVERSE REACTIONS The following adverse reactions are discussed in greater detail in other sections of the label. Hypersensitivity [see Contraindications (4)] Suicidal Thoughts and Behaviors in Adolescents and Young Adults [see Warnings and Precautions (5.1)] Serotonin Syndrome [see Warnings and Precautions (5.2)] Elevated Blood Pressure [see Warnings and Precautions (5.3)] Elevated Heart Rate [see Warnings and Precautions (5.4)] Abnormal Bleeding [see Warnings and Precautions (5.5)] Angle Closure Glaucoma [see Warnings and Precautions (5.6)] Urinary Hesitation or Retention [see Warnings and Precautions (5.7)] Activation of Mania/Hypomania [see Warnings and Precautions (5.8)] Seizure [see Warnings and Precautions (5.9)] Discontinuation Syndrome [see Warnings and Precautions (5.10)] Hyponatremia [see Warnings and Precautions (5.11)] 6.1 Clinical Studies Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice. Patient exposure The safety of FETZIMA was evaluated in 2,673 patients (18-78 years of age) diagnosed with MDD who participated in clinical studies, representing 942 patient-years of exposure. Among the 2,673 FETZIMA-treated patients, 1,583 were exposed to FETZIMA in short-term, placebo-controlled studies. There were 825 patients who continued from short-term studies into a one-year, open-label extension study. Of the 2,673 patients exposed to at least one dose of FETZIMA, 737 patients were exposed to FETZIMA for at least 6 months and 367 were exposed for one year. In these studies FETZIMA was given at doses ranging from 40-120 mg once daily and was given without regard to food. Adverse reactions reported as reasons for discontinuation of treatment In the short-term placebo-controlled pre-marketing studies for MDD, 9% of the 1,583 patients who received FETZIMA (40-120 mg) discontinued treatment due to an adverse event, compared with 3% of the 1,040 placebo-treated patients in those studies. The most common adverse reaction leading to discontinuation in at least 1% of the FETZIMA-treated patients in the short-term placebo-controlled studies was nausea (1.5%). Common adverse reactions in placebo-controlled MDD studies The most commonly observed adverse events in FETZIMA-treated MDD patients in placebo-controlled studies (incidence ≥ 5% and at least twice the rate of placebo) were: nausea, constipation, hyperhidrosis, heart rate increased, erectile dysfunction, tachycardia, vomiting, and palpitations. Table 3 shows the incidence of adverse reactions that occurred in ≥ 2% of FETZIMA-treated MDD patients and at least twice the rate of placebo in the placebo-controlled studies. Table 3 Adverse Reactions Occurring in ≥ 2% of FETZIMA-treated Patients and at Least Twice the rate of Placebo-treated Patients
b Percentage is relative to the number of patients in the associated demographic sex category. Fewer than 2% of FETZIMA-treated MDD female patients in placebo-controlled clinical studies reported adverse events related to sexual function. c erectile dysfunction includes: erectile dysfunction, organic erectile dysfunction and psychogenic erectile dysfunction d testicular pain includes: testicular pain, epididymitis, and seminal vesiculitis e ejaculation disorder includes: ejaculation disorder, ejaculation delayed, ejaculation failure, and premature ejaculation f Heart rate increased also includes: orthostatic heart rate response increased g Blood pressure increased also includes: blood pressure systolic increased, blood pressure diastolic increased and blood pressure orthostatic increased h Rash also includes: rash generalized, rash maculo-papular, rash erythematous and rash macular i Hypotension also includes: orthostatic hypotension and dizziness postural j Hypertension also includes: labile hypertension N = number of patients in the Safety Population Dose-related adverse reactions In pooled data from the short-term placebo-controlled fixed-dose studies, there were no dose-related adverse reactions (greater than 2% overall incidence) in patients treated with FETZIMA across the dose range 40-120 mg once daily, with the exception of erectile dysfunction and urinary hesitation (see Table 4). Table 4 Dose-Related Adverse Reactions
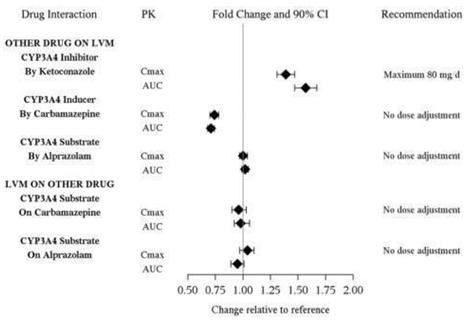
N = number of patients in the Safety Population Other adverse reactions observed in clinical studies Other infrequent adverse reactions, not described elsewhere in the label, occurring at an incidence of < 2% in MDD patients treated with FETZIMA were: Cardiac disorders: Angina pectoris; Supraventricular and Ventricular extrasystoles Eye disorders: Dry eye; Vision blurred; Conjunctival hemorrhage General disorders: Chest pain; Thirst Gastrointestinal disorders: Abdominal pain; Flatulence Investigations disorders: Blood cholesterol increased; Liver function test abnormal Nervous System disorders: Migraine; Paraesthesia; Syncope; Extrapyramidal disorder Psychiatric disorders: Agitation; Anger; Bruxism; Panic attack; Tension; Aggression Renal and Urinary disorder: Pollakiuria; Hematuria; Proteinuria Respiratory, thoracic and mediastinal disorders: Yawning Skin and subcutaneous tissue disorders: Dry skin; Pruritus; Urticaria 7 DRUG INTERACTIONS Other than CYP3A4 drug interactions, FETZIMA is predicted, based on in vitro studies, to have a low potential to be involved in clinically significant pharmacokinetic drug interactions. 7.1 Monoamine Oxidase Inhibitors (MAOIs) see Dosage and Administration (2.5, 2.6), Contraindications (4), and Warnings and Precautions (5.2)] 7.2 Serotonergic Drugs see Dosage and Administration (2.5, 2.6), Contraindications (4), and Warnings and Precautions (5.2)] 7.3 Drugs that Interfere with Hemostasis (e.g., NSAIDs, Aspirin, and Warfarin) Serotonin release by platelets plays an important role in hemostasis. Epidemiological studies of case-control and cohort design have demonstrated an association between use of psychotropic drugs that interfere with serotonin reuptake and the occurrence of upper gastrointestinal bleeding. These studies have also shown that concurrent use of an NSAID or aspirin may potentiate this risk of bleeding. Altered anticoagulant effects, including increased bleeding, have been reported when SSRIs and SNRIs are co-administered with warfarin. Patients receiving warfarin therapy should be carefully monitored when FETZIMA is initiated or discontinued [see Warnings and Precautions (5.5)]. 7.4 Potential for Other Drugs to Affect FETZIMA Dose adjustment is recommended when FETZIMA is co-administered with strong inhibitors of CYP3A4 (e.g. ketoconazole) [see Dosage and Administration (2.7)]. An in vivo study showed a clinically meaningful increase in levomilnacipran exposure when FETZIMA was co-administered with the CYP3A4 inhibitor ketoconazole (see Figure 1). No dose adjustment of FETZIMA is needed when co-administered with a CYP3A4 inducer or substrate. In vivo studies showed no clinically meaningful change in levomilnacipran exposure when co-administered with the CYP3A4 inducer carbamazepine or the CYP3A4 substrate alprazolam (see Figure 1). No dose adjustment of FETZIMA is needed when co-administered with inhibitors of CYP2C8 , CYP2C19, CYP2D6, CYP2J2, P-glycoprotein, BCRP, OATP1B1, OATP1B3, OAT1, OAT3, or OCT2. In vitro studies suggested that CYP2C8, CYP2C19, CYP2D6, and CYP2J2 had minimal contributions to metabolism of levomilnacipran. In addition, levomilnacipran is not a substrate of BCRP, OATP1B1, OATP1B3, OAT1, OAT3, or OCT2 and is a weak substrate of P-gp. Figure 1 PK Interactions between Levomilnacipran (LVM) and Other Drugs
a Difference (drug minus placebo) in least-squares mean change from baseline to endpoint (Week 8). Doses statistically significantly superior to placebo. Post-hoc analyses of the relationships between treatment outcome and age, gender, and race did not suggest any differential responsiveness on the basis of these patient characteristics. 16 HOW SUPPLIED/STORAGE AND HANDLING FETZIMA extended-release capsules are supplied in the following configurations: 0456-2240-30 bottle of 30 0456-2240-90 bottle of 90 0456-2240-63 UD 100 NDC 69189-2241-1 single dose pack with 1 capsule as repackaged by Avera McKennan Hospital Storage and Handling All package configurations: Store at 25°C (77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [See USP Controlled Room Temperature]. 17 PATIENT COUNSELING INFORMATION See FDA-approved patient labeling (Medication Guide) Information for Patients Advise patients, their families, and their caregivers about the benefits and risks associated with treatment with FETZIMA and counsel them on its appropriate use. Advise patients, their families, and their caregivers to read the Medication Guide and assist them in understanding its contents. The complete text of the Medication Guide is reprinted at the end of this document. Suicide Risk Advise patients and caregivers to look for the emergence of suicidality, especially early during treatment and when the dose is adjusted up or down [see Box Warning and Warnings and Precautions (5.1)]. Dosing and Administration Advise patients that FETZIMA should be swallowed whole and should not be chewed, crushed or opened. Advise patients that FETZIMA can be taken with or without food. FETZIMA should be initiated with a dose of 20 mg once daily for 2 days and then increased to 40 mg once daily. Based on efficacy and tolerability, FETZIMA may then be increased in increments of 40 mg at intervals of 2 or more days. The maximum recommended dose is 120 mg once daily. Instruct patients if they miss a dose, to take the missed dose as soon as they remember. If it is almost time for the next dose, instruct them to skip the missed dose and take their next dose at the regular time. Advise them not to take two doses of FETZIMA at the same time. Concomitant Medication Instruct patients not to take FETZIMA with an MAOI or within 14 days of stopping an MAOI and to allow 7 days after stopping FETZIMA before starting an MAOI [see Contraindications (4)]. Serotonin Syndrome Caution patients about the risk of serotonin syndrome, particularly with the concomitant use of FETZIMA and triptans, tramadol, tryptophan supplements, other serotonergic agents, or antipsychotic drugs [see Warnings and Precautions (5.2) and Drug Interactions (7.2)]. Effect on Blood Pressure and Heart Rate Advise patients that they should have regular monitoring of blood pressure and heart rate when taking FETZIMA [see Warnings and Precautions (5.3, 5.4)]. Abnormal Bleeding Caution patients about the concomitant use of FETZIMA and NSAIDs, aspirin, warfarin, or other drugs that affect coagulation since combined use of psychotropic drugs that interfere with serotonin reuptake and these agents has been associated with an increased risk of abnormal bleeding [see Warnings and Precautions (5.5)]. Angle Closure Glaucoma Patients should be advised that taking FETZIMA can cause mild pupillary dilation, which in susceptible individuals, can lead to an episode of angle closure glaucoma. Pre-existing glaucoma is almost always open-angle glaucoma because angle closure glaucoma, when diagnosed, can be treated definitively with iridectomy. Open-angle glaucoma is not a risk factor for angle closure glaucoma. Patients may wish to be examined to determine whether they are susceptible to angle closure, and have a prophylactic procedure (e.g., iridectomy), if they are susceptible [see Warnings and Precautions (5.6)]. Urinary Hesitation or Retention Caution patients about the risk of urinary hesitation and retention while taking FETZIMA, particularly in patients prone to obstructive urinary disorders [see Warnings and Precautions (5.7)]. Activation of Mania/Hypomania Advise patients and their caregivers to observe for signs of activation of mania/hypomania [see Warnings and Precautions (5.8)]. Seizures Caution patients about using FETZIMA if they have a history of a seizure disorder [see Warnings and Precautions (5.9)]. Patients with a history of seizures were excluded from clinical studies. Discontinuation Syndrome Advise patients not to stop taking FETZIMA without first talking with their healthcare provider. Patients should be aware that discontinuation effects may occur when suddenly stopping FETZIMA [see Warnings and Precautions (5.10)]. Hyponatremia Advise patients that if they are treated with diuretics, or are otherwise volume depleted, or are elderly, they may be at greater risk of developing hyponatremia while taking FETZIMA [see Warnings and Precautions (5.11)]. Alcohol Advise patients to avoid consumption of alcohol while taking FETZIMA [see Drug Interactions (7.6)]. Allergic Reactions Advise patients to notify their healthcare provider if they develop an allergic reaction such as rash, hives, swelling, or difficulty breathing. Pregnancy Advise patients to notify their healthcare provider if they become pregnant or intend to become pregnant during therapy with FETZIMA [see Use in Specific Populations (8.1)]. Nursing Mothers Advise patients to notify their healthcare provider if they are breastfeeding an infant and would like to continue or start FETZIMA [see Use in Specific Populations (8.3)]. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=6f746bcd-b121-447a-9f5d-c9391e886159 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||