|
肺结核新药SIRTURO(BEDAQUILINE FUMARATE)TABLET ORAL-120周跟踪结果发表,显著提高治愈率
HIGHLIGHTS OF PRESCRIBING INFORMATION
SIRTURO is a diarylquinoline antimycobacterial drug indicated as part of combination therapy in adults (18 years and older) with pulmonary multi-drug resistant tuberculosis (MDR-TB). Reserve SIRTURO for use when an effective treatment regimen cannot otherwise be provided. Administer SIRTURO by directly observed therapy (DOT). (1, 2.1) This indication is approved under accelerated approval based on time to sputum culture conversion. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. (1, 14) Limitations of Use: Do not use SIRTURO for the treatment of latent, extra-pulmonary or drug-sensitive tuberculosis or for the treatment of infections caused by non-tuberculous mycobacteria (1). Safety and efficacy of SIRTURO in HIV-infected patients with MDR-TB have not been established, as clinical data are limited (14). DOSAGE AND ADMINISTRATION Emphasize need for compliance with full course of therapy (2.1) Prior to administration, obtain ECG, liver enzymes and electrolytes. Obtain susceptibility information for the background regimen against Mycobacterium tuberculosis isolate if possible. (2.2) Only use SIRTURO in combination with at least 3 other drugs to which the patient's MDR-TB isolate has been shown to be susceptible in vitro. If in vitro testing results are unavailable, may initiate SIRTURO in combination with at least 4 other drugs to which patient's MDR-TB isolate is likely to be susceptible (2.3) Recommended dosage: 400 mg once daily for 2 weeks followed by 200 mg 3 times per week (with at least 48 hours between doses) for 22 weeks (2.3) Swallow SIRTURO tablets whole with water and take with food. (2.3) DOSAGE FORMS AND STRENGTHS Tablets: 100 mg (3) CONTRAINDICATIONS None. (4) WARNINGS AND PRECAUTIONS QT prolongation can occur with SIRTURO. Monitor ECGs and discontinue SIRTURO if significant ventricular arrhythmia or QTcF interval > 500 ms develops. (5.2) Hepatotoxicity may occur with use of SIRTURO. Monitor liver-related laboratory tests. Discontinue if evidence of liver injury. (5.3) ADVERSE REACTIONS The most common adverse reactions reported in 10% or more of patients treated with SIRTURO were nausea, arthralgia, headache, hemoptysis and chest pain. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Janssen Therapeutics, Division of Janssen Products, LP at 1-800-JANSSEN (1-800-526-7736) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS Avoid use of strong and moderate CYP3A4 inducers with SIRTURO. (7.1, 7.3) Avoid use for more than 14 consecutive days of systemic strong CYP3A4 inhibitors with SIRTURO unless the benefit outweighs the risk. Monitor for SIRTURO-related adverse reactions. (7.1) USE IN SPECIFIC POPULATIONS Use with caution in patients with severe hepatic impairment and only when the benefits outweigh the risks. Monitor for SIRTURO-related adverse reactions. (8.6) Use with caution in patients with severe renal impairment. (8.7) See 17 for PATIENT COUNSELING INFORMATION and Medication Guide. Revised: 12/2015 FULL PRESCRIBING INFORMATION: CONTENTS* 1 INDICATIONS AND USAGE SIRTURO is a diarylquinoline antimycobacterial drug indicated as part of combination therapy in the treatment of adults (18 years and older) with pulmonary multi-drug resistant tuberculosis (MDR-TB). Reserve SIRTURO for use when an effective treatment regimen cannot otherwise be provided. Administer SIRTURO by directly observed therapy (DOT). This indication is approved under accelerated approval based on time to sputum culture conversion [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. Limitations of Use: Do not use SIRTURO for the treatment of: Latent infection due to Mycobacterium tuberculosis Drug-sensitive tuberculosis Extra-pulmonary tuberculosis Infections caused by non-tuberculous mycobacteria The safety and efficacy of SIRTURO in the treatment of HIV infected patients with MDR-TB have not been established as clinical data are limited [see Clinical Studies (14)]. 2 DOSAGE AND ADMINISTRATION 2.1 Important Administration Instructions Administer SIRTURO by directly observed therapy (DOT). Use SIRTURO only in combination with other anti-mycobacterial drugs [see Dosage and Administration (2.3)]. Emphasize the need for compliance with full course of therapy. 2.2 Required Testing Prior to Administration Prior to treatment with SIRTURO, obtain the following: Susceptibility information for the background regimen against M. tuberculosis isolate if possible [see Dosage and Administration (2.3)] ECG [see Warnings and Precautions (5.2)] Serum potassium, calcium, and magnesium concentrations [see Warnings and Precautions (5.2)] Liver enzymes [see Warnings and Precautions (5.3)] 2.3 Recommended Dosage in Combination Therapy Only use SIRTURO in combination with at least 3 other drugs to which the patient's MDR-TB isolate has been shown to be susceptible in vitro. If in vitro testing results are unavailable, SIRTURO treatment may be initiated in combination with at least 4 other drugs to which the patient's MDR-TB isolate is likely to be susceptible. Refer to the prescribing information of the drugs used in combination with SIRTURO. The recommended dosage of SIRTURO is 400 mg orally once daily for the first two weeks, followed by 200 mg orally three times per week (with at least 48 hours between doses) for 22 weeks (total duration of 24 weeks). The SIRTURO tablet should be swallowed whole with water and taken with food. If a dose is missed during the first 2 weeks of treatment, do not administer the missed dose (skip the dose and then continue the daily dosing regimen). From Week 3 onwards, if a 200 mg dose is missed, administer the missed dose as soon as possible, and then resume the 3 times a week dosing regimen. 3 DOSAGE FORMS AND STRENGTHS SIRTURO tablets, 100 mg are uncoated white to almost white round biconvex with debossing of "T" over "207" on one side and "100" on the other side. 4 CONTRAINDICATIONS None. 5 WARNINGS AND PRECAUTIONS 5.1 Increased Mortality An increased risk of death was seen in the SIRTURO treatment group (9/79, 11.4%) compared to the placebo treatment group (2/81, 2.5%) in one placebo-controlled trial (based on the 120-week visit window). One death occurred during the 24 weeks of administration of SIRTURO. The imbalance in deaths is unexplained. No discernible pattern between death and sputum culture conversion, relapse, sensitivity to other drugs used to treat tuberculosis, HIV status, or severity of disease could be observed. Only use SIRTURO when an effective treatment regimen cannot otherwise be provided [see Adverse Reactions (6)]. 5.2 QT Prolongation SIRTURO prolongs the QT interval. Obtain an ECG before initiation of treatment, and at least 2, 12, and 24 weeks after starting treatment with SIRTURO. Obtain serum potassium, calcium, and magnesium at baseline and correct if abnormal. Monitor electrolytes if QT prolongation is detected [see Adverse Reactions (6.1) and Drug Interactions (7.4)]. SIRTURO has not been studied in patients with ventricular arrhythmias or recent myocardial infarction. The following may increase the risk for QT prolongation when patients are receiving SIRTURO: use with other QT prolonging drugs including fluoroquinolones and macrolide antibacterial drugs and the antimycobacterial drug, clofazimine a history of Torsade de Pointes a history of congenital long QT syndrome a history of or ongoing hypothyroidism a history of or ongoing bradyarrhythmias a history of uncompensated heart failure serum calcium, magnesium, or potassium levels below the lower limits of normal If necessary, bedaquiline treatment initiation could be considered in these patients after a favorable benefit risk assessment and with frequent ECG monitoring. Discontinue SIRTURO and all other QT prolonging drugs if the patient develops: Clinically significant ventricular arrhythmia A QTcF interval of greater than 500 ms (confirmed by repeat ECG) If syncope occurs, obtain an ECG to detect QT prolongation. 5.3 Hepatotoxicity More hepatic-related adverse reactions were reported with the use of SIRTURO plus other drugs used to treat tuberculosis compared to other drugs used to treat tuberculosis without the addition of SIRTURO. Alcohol and other hepatotoxic drugs should be avoided while on SIRTURO, especially in patients with impaired hepatic function. Monitor symptoms (such as fatigue, anorexia, nausea, jaundice, dark urine, liver tenderness and hepatomegaly) and laboratory tests (ALT, AST, alkaline phosphatase, and bilirubin) at baseline, monthly while on treatment, and as needed. Test for viral hepatitis and discontinue other hepatotoxic medications if evidence of new or worsening liver dysfunction occurs. Discontinue SIRTURO if: aminotransferase elevations are accompanied by total bilirubin elevation greater than two times the upper limit of normal aminotransferase elevations are greater than eight times the upper limit of normal aminotransferase elevations are greater than five times the upper limit of normal and persist beyond two weeks 5.4 Drug Interactions CYP3A4 inducers/inhibitors Bedaquiline is metabolized by CYP3A4 and its systemic exposure and therapeutic effect may therefore be reduced during co-administration with inducers of CYP3A4. Avoid co-administration of strong CYP3A4 inducers, such as rifamycins (i.e., rifampin, rifapentine and rifabutin), or moderate CYP3A4 inducers, such as efavirenz, during treatment with SIRTURO [see Drug Interactions (7.1)]. Co-administration of SIRTURO with strong CYP3A4 inhibitors may increase the systemic exposure to bedaquiline, which could potentially increase the risk of adverse reactions. Therefore, avoid the use of strong CYP3A4 inhibitors for more than 14 consecutive days while on SIRTURO, unless the benefit of treatment with the drug combination outweighs the risk [see Drug Interactions (7.1)]. Appropriate clinical monitoring for SIRTURO-related adverse reactions is recommended. 6 ADVERSE REACTIONS The following serious adverse reactions are discussed elsewhere in the labeling: Increased mortality [see Warnings and Precautions (5.1)] QT Prolongation [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.2)] Hepatotoxicity [see Warnings and Precautions (5.3)] Drug Interactions [see Warnings and Precautions (5.4)] 6.1 Clinical Studies Experience Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to the rates in the clinical studies of another drug and may not reflect the rates observed in clinical practice. Use SIRTURO only in combination with other anti-mycobacterial drugs [see Dosage and Administration (2.3)]. Refer to the prescribing information of the drugs used in combination with SIRTURO for their respective adverse reactions. Adverse drug reactions for SIRTURO were identified from the pooled safety data from 335 SIRTURO-exposed patients who received 8 weeks (Study 2) and 24 weeks (Studies 1 and 3) at the proposed dose. Studies 1 and 2 were randomized, double-blind, placebo-controlled trial in newly diagnosed patients with pulmonary MDR-TB. In both treatment arms, patients received SIRTURO or placebo in combination with other drugs used to treat MDR-TB. Study 3 was an open-label, noncomparative study with SIRTURO administered as part of an individualized pulmonary MDR-TB treatment regimen in previously treated patients. In Study 1, 35% were Black, 17.5% were Hispanic, 12.5% were White, 9.4% were Asian, and 25.6% were of another race. Eight of 79 (10.1%) patients in the SIRTURO group and 16 of 81 (19.8%) patients in the placebo treatment group were HIV-infected. Seven (8.9%) SIRTURO-treated patients and six (7.4%) placebo-treated patients discontinued Study 1 because of an adverse reaction. Table 1: Select Adverse Reactions from Study 1 That Occurred More Frequently Than Placebo During Treatment with SIRTURO
No additional unique Adverse Reactions were identified from the uncontrolled Study 3. In both Studies 1 and 2, aminotransferase elevations of at least 3 times the upper limit of normal developed more frequently in the SIRTURO treatment group (11/102 [10.8%] vs 6/105 [5.7%]) than in the placebo treatment group. In Study 3, 22/230 (9.6%) patients had alanine aminotransferase or aspartate aminotransferase greater than or equal to 3 times the upper limit of normal during the overall treatment period. Increased Mortality In Study 1, there was a statistically significant increased mortality risk by Week 120 in the SIRTURO treatment group compared to the placebo treatment group (9/79 (11.4%) versus 2/81 (2.5%), p-value=0.03, an exact 95% confidence interval of the difference [1.1%, 18.2%]). Five of the 9 SIRTURO deaths and the 2 placebo deaths were tuberculosis-related. One death occurred during the 24-week SIRTURO treatment period. The median time to death for the remaining eight subjects in the SIRTURO treatment group was 329 days after last intake of SIRTURO. The imbalance in deaths is unexplained; no discernible pattern between death and sputum conversion, relapse, sensitivity to other drugs used to treat tuberculosis, HIV status, and severity of disease was observed. In the open-label Study 3, 6.9% (16/233) subjects died. The most common cause of death as reported by the investigator was TB (9 subjects). All but one subject who died of TB had not converted or had relapsed. The causes of death in the remaining subjects varied. 7 DRUG INTERACTIONS 7.1 CYP3A4 Inducers/Inhibitors Bedaquiline exposure may be reduced during co-administration with inducers of CYP3A4 and increased during co-administration with inhibitors of CYP3A4. CYP3A4 Inducers Due to the possibility of a reduction of the therapeutic effect of bedaquiline because of the decrease in systemic exposure, co-administration of strong CYP3A4 inducers, such as rifamycins (i.e., rifampin, rifapentine and rifabutin), or moderate CYP3A4 inducers should be avoided during treatment with SIRTURO [see Clinical Pharmacology (12.3)]. CYP3A4 inhibitors Due to the potential risk of adverse reactions to bedaquiline because of the increase in systemic exposure, prolonged co-administration of bedaquiline and strong CYP3A4 inhibitors, such as ketoconazole or itraconazole, for more than 14 consecutive days should be avoided unless the benefit outweighs the risk [see Clinical Pharmacology (12.3)]. Appropriate clinical monitoring for SIRTURO-related adverse reactions is recommended. 7.2 Other Antimicrobial Medications No dose-adjustment of isoniazid or pyrazinamide is required during co-administration with SIRTURO. In a placebo-controlled clinical trial in patients with MDR-TB, no major impact of co-administration of SIRTURO on the pharmacokinetics of ethambutol, kanamycin, pyrazinamide, ofloxacin or cycloserine was observed. 7.3 Antiretroviral Medications Lopinavir/ritonavir Although clinical data in HIV/MDR-TB co-infected patients on the combined use of lopinavir (400 mg)/ritonavir (100 mg) with SIRTURO are not available, use SIRTURO with caution when co-administered with lopinavir/ritonavir and only if the benefit outweighs the risk [see Clinical Pharmacology (12.3)]. Nevirapine No dosage adjustment of bedaquiline is required when co-administered with nevirapine [see Clinical Pharmacology (12.3)]. Efavirenz Concomitant administration of bedaquiline and efavirenz, or other moderate CYP3A inducers, should be avoided [see Warnings and Precautions (5.4)]. 7.4 QT Interval Prolonging Drugs In a drug interaction study of bedaquiline and ketoconazole, a greater effect on QTc was observed after repeated dosing with bedaquiline and ketoconazole in combination than after repeated dosing with the individual drugs. Additive or synergistic QT prolongation was observed when bedaquiline was co-administered with other drugs that prolong the QT interval. In Study 3, mean increases in QTc were larger in the 17 subjects who were taking clofazimine with bedaquiline at Week 24 (mean change from reference of 31.9 ms) than in subjects who were not taking clofazimine with bedaquiline at Week 24 (mean change from baseline of 12.3 ms). Monitor ECGs if SIRTURO is co-administered to patients receiving other drugs that prolong the QTc interval, and discontinue SIRTURO if evidence of serious ventricular arrhythmia or QTcF interval greater than 500 ms. [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.2)]. 8 USE IN SPECIFIC POPULATIONS 8.1 Pregnancy Pregnancy Category B Reproduction studies performed in rats and rabbits have revealed no evidence of harm to the fetus due to bedaquiline. In these studies, the corresponding plasma exposure (AUC) was 2-fold higher in rats compared to humans. There are, however, no adequate and well-controlled studies of SIRTURO in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed. 8.3 Nursing Mothers It is not known whether bedaquiline or its metabolites are excreted in human milk, but rat studies have shown that drug is concentrated in breast milk. In rats, treated with bedaquiline at doses 1 time to 2 times the clinical dose (based on AUC comparisons), concentrations in milk were 6-fold to 12-fold higher than the maximum concentration observed in maternal plasma. Pups from these dams showed reduced body weights compared to control animals throughout the lactation period. Because of the potential for adverse reactions in nursing infants, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. 8.4 Pediatric Use The safety and effectiveness of SIRTURO in pediatric patients have not been established. 8.5 Geriatric Use Because of limited data, differences in outcomes or specific risks with SIRTURO cannot be ruled out for patients 65 years of age and older. 8.6 Hepatic Impairment The pharmacokinetics of bedaquiline were assessed after single-dose administration to subjects with moderate hepatic impairment (Child-Pugh B) [see Clinical Pharmacology (12.3)]. Based on these results, no dose adjustment is necessary for SIRTURO in patients with mild or moderate hepatic impairment. SIRTURO has not been studied in patients with severe hepatic impairment and should be used with caution in these patients only when the benefits outweigh the risks. Clinical monitoring for SIRTURO-related adverse reactions is recommended [see Warnings and Precautions (5.3)]. 8.7 Renal Impairment SIRTURO has mainly been studied in patients with normal renal function. Renal excretion of unchanged bedaquiline is not substantial (less than or equal to 0.001%). No dose adjustment is required in patients with mild or moderate renal impairment. In patients with severe renal impairment or end stage renal disease requiring hemodialysis or peritoneal dialysis, SIRTURO should be used with caution [see Clinical Pharmacology (12.3)]. Monitor for adverse reactions of SIRTURO when administered to patients with severe renal impairment or end stage renal disease requiring hemodialysis or peritoneal dialysis. 10 OVERDOSAGE There is no experience with the treatment of acute overdose with SIRTURO. Take general measures to support basic vital functions including monitoring of vital signs and ECG (QT interval) in case of deliberate or accidental overdose. Removal of unabsorbed bedaquiline may be achieved by the administration of activated charcoal. Since bedaquiline is highly protein-bound, dialysis is not likely to significantly remove bedaquiline from plasma. 11 DESCRIPTION SIRTURO (bedaquiline) for oral administration is available as 100 mg strength tablets. Each tablet contains 120.89 mg of bedaquiline fumarate drug substance, which is equivalent to 100 mg of bedaquiline. Bedaquiline is a diarylquinoline antimycobacterial drug. Bedaquiline fumarate is a white to almost white powder and is practically insoluble in aqueous media. The chemical name of bedaquiline fumarate is (1R, 2S)-1-(6-bromo-2-methoxy-3-quinolinyl)-4-(dimethylamino)-2-(1-naphthalenyl)-1-phenyl-2-butanol compound with fumaric acid (1:1). It has a molecular formula of C32H31BrN2O2∙C4H4O4 and a molecular weight of 671.58 (555.50 + 116.07). The molecular structure of bedaquiline fumarate is the following:
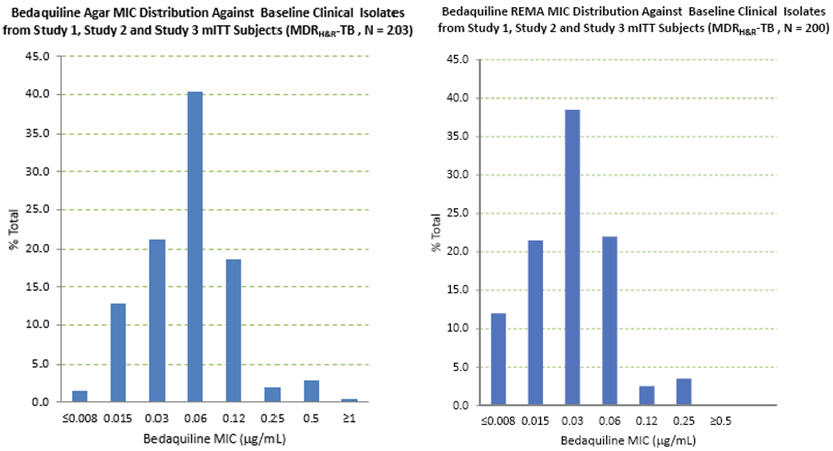
Nineteen patients in the efficacy population of study 3 had bedaquiline susceptibility testing results of paired (baseline and post-baseline, all of which were at Week 24 or later) genotypically identical isolates. Twelve of the 19 had a post-baseline ≥4-fold increase in bedaquiline MIC. Whole genome sequencing of 9 of these 12 post-baseline isolates was done and no mutations were found in the ATP synthase operon. All 9 were found to have a mutation in Rv0678. Eleven of the twelve (11/12) increases in bedaquiline MIC were seen in patients with pre-XDR-TB or with XDR-TB. Pre-XDR-TB is defined as MDR-TB isolates resistant to either a fluoroquinolone or a second line injectable drug, and XDR-TB as MDR-TB isolates resistant to both a fluoroquinolone and a second line injectable drug. Based on available data, response rate (culture conversion at week 120 endpoint) was similar in subjects with ≥4-fold increases in bedaquiline MIC (5/12) and subjects with < 4-fold increases (3/7). Quality Control Susceptibility test procedures require the use of laboratory controls to monitor and ensure the accuracy and precision of testing. Assays using standard bedaquiline powder should provide the following range of MIC values shown in Table 3. Table 3: Quality Control Ranges using Agar and Broth Dilution Methods and M. tuberculosis H37Rv
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility Bedaquiline was not carcinogenic in rats up to the maximum tolerated dose of 10 mg/kg/day. Exposures at this dose in rats (AUCs) were within 1-fold to 2-fold of those observed in subjects in the Phase 2 clinical trials. No mutagenic or clastogenic effects were detected in the in vitro non-mammalian reverse mutation (Ames) test, in vitro mammalian (mouse lymphoma) forward mutation assay and an in vivo mouse bone marrow micronucleus assay. SIRTURO had no effects on fertility when evaluated in male and female rats. No relevant drug-related effects on developmental toxicity parameters were observed in rats and rabbits. The corresponding plasma exposure (AUC) was 2-fold higher in rats and lower for rabbits compared to humans. There was no effect of maternal treatment with bedaquiline at any dose level on sexual maturation, behavioral development, mating performance, fertility or reproductive capacity of the F1 generation animals. Body weight decreases in pups were noted in high dose groups during the lactation period after exposure to bedaquiline via milk and were not a consequence of in utero exposure. Concentrations of bedaquiline in milk were 6-fold to 12-fold higher that the maximum concentration observed in maternal plasma. 13.2 Animal Toxicology and/or Pharmacology Bedaquiline is a cationic, amphiphilic drug that induced phospholipidosis (at almost all doses, even after very short exposures) in drug-treated animals, mainly in cells of the monocytic phagocytic system (MPS). All species tested showed drug-related increases in pigment-laden and/or foamy macrophages, mostly in the lymph nodes, spleen, lungs, liver, stomach, skeletal muscle, pancreas and/or uterus. After treatment ended, these findings were slowly reversible. Muscle degeneration was observed in several species at the highest doses tested. For example the diaphragm, esophagus, quadriceps and tongue of rats were affected after 26 weeks of treatment at doses similar to clinical exposures based on AUC comparisons. These findings were not seen after a 12-week, treatment-free, recovery period and were not present in rats given the same dose biweekly. Degeneration of the fundic mucosa of the stomach, hepatocellular hypertrophy and pancreatitis were also seen. 14 CLINICAL STUDIES A placebo-controlled, double-blind, randomized trial (Study 1) was conducted in patients with newly diagnosed sputum smear-positive MDR pulmonary M. tuberculosis. All patients received a combination of five other antimycobacterial drugs used to treat MDR-TB (i.e., ethionamide, kanamycin, pyrazinamide, ofloxacin, and cycloserine/terizidone or available alternative) for a total duration of 18–24 months or at least 12 months after the first confirmed negative culture. In addition to this regimen, patients were randomized to receive 24 weeks of treatment with SIRTURO 400 mg once daily for the first 2 weeks followed by 200 mg 3 times per week for 22 weeks or matching placebo for the same duration. Overall, 79 patients were randomized to the SIRTURO arm and 81 to the placebo arm. A final evaluation was conducted at Week 120. Sixty-seven patients randomized to SIRTURO and 66 patients randomized to placebo had confirmed MDR-TB, based on susceptibility tests (taken prior to randomization) or medical history if no susceptibility results were available, and were included in the efficacy analyses. Demographics were as follows: 63% of the study population was male, with a median age of 34 years, 35% were Black, and 15% were HIV-positive (median CD4 cell count 468 cells/µL). Most patients had cavitation in one lung (62%); and 18% of patients had cavitation in both lungs. Time to sputum culture conversion was defined as the interval in days between the first dose of study drug and the date of the first of two consecutive negative sputum cultures collected at least 25 days apart during treatment. In this trial, the SIRTURO treatment group had a decreased time to culture conversion and improved culture conversion rates compared to the placebo treatment group at Week 24. Median time to culture conversion was 83 days for the SIRTURO treatment group compared to 125 days for the placebo treatment group. Table 4 shows the proportion of patients with sputum culture conversion at Week 24 and Week 120. Table 4: Culture Conversion Status in Patients with MDR-TB at Week 24 and Week 120 in Study 1
Patients received 24 weeks of SIRTURO or placebo for the first 24 weeks and received a combination of other antimycobacterial drugs for up to 96 weeks. Study 2 was a smaller placebo controlled study designed similarly to Study 1 except that SIRTURO or placebo was given for only 8 weeks instead of 24 weeks. Patients were randomized to either SIRTURO and other drugs used to treat MDR-TB (SIRTURO treatment group) (n=23) or placebo and other drugs used to treat MDR-TB (placebo treatment group) (n=24). Twenty-one patients randomized to the SIRTURO treatment group and 23 patients randomized to the placebo treatment group had confirmed MDR-TB based on subjects' baseline M. tuberculosis isolate obtained prior to randomization. The SIRTURO treatment group had a decreased time to culture conversion and improved culture conversion rates compared to the placebo treatment group at Week 8. At Weeks 8 and 24, the differences in culture conversion proportions were 38.9% (95% CI: [12.3%, 63.1%] and p-value: 0.004), 15.7% (95% CI: [-11.9%, 41.9%] and p-value: 0.32), respectively. Study 3 was a Phase 2b, uncontrolled study to evaluate the safety, tolerability, and efficacy of SIRTURO as part of an individualized MDR-TB treatment regimen in 233 patients with sputum smear positive (within 6 months prior to screening) pulmonary MDR-TB. Patients received SIRTURO for 24 weeks in combination with antibacterial drugs. Upon completion of the 24 week treatment with SIRTURO, all patients continued to receive their background regimen in accordance with national TB program (NTP) treatment guidelines. A final evaluation was conducted at Week 120. Treatment responses to SIRTURO at week 120 were generally consistent with those from Study 1. 15 REFERENCES 1.Clinical and Laboratory Standards Institute (CLSI). Susceptibility Testing of Mycobacteria, Nocardiaceae, and other Aerobic Actinomycetes; Approved Standard – Second Edition. CLSI document M24-A2. Clinical and Laboratory Standards Institute, 950 West Valley Rd., Suite 2500, Wayne, PA, 19087, 2011. 2.Martin A, Portaels F, Palomino JC. Colorimetric redox-indicator methods for the rapid detection of multidrug resistance in Mycobacterium tuberculosis: a systematic review and meta-analysis. J Antimicrob Chemother. 2007; 59 (2): 175-83. 3.Clinical and Laboratory Institute Standards (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard — Nineth Edition. CLSI Document M07-A9. Clinical and Laboratory Standards Institute, 950 West Valley Rd., Suite 2500, Wayne, PA, 19087, 2012. 16 HOW SUPPLIED/STORAGE AND HANDLING How supplied SIRTURO is supplied as uncoated white to almost white round biconvex 100 mg tablets with debossing of "T" over "207" on one side and "100" on the other side. The tablets are packaged in white high density polyethylene (HDPE) bottles with child-resistant polypropylene (PP) closure with induction seal liner. Each bottle contains 188 tablets. NDC 59676-701-01 Storage and handling Keep out of reach of children. Dispense in original container. Store tablets dispensed outside the original container in a tight light-resistant container with an expiration date not to exceed 3 months. Store at 25°C (77°F); excursions permitted to 15–30°C (59–86°F). [See USP Controlled Room Temperature]
https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=1534c9ae-4948-4cf4-9f66-222a99db6d0e | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||