|
英文药名:XALKORI(Crizotinib)caps 中文药名:克唑替尼胶囊 生产厂家:Pfizer
a. Except lymphopenia (unless associated with clinical events, e.g., opportunistic infections).
a. NCI Common Terminology Criteria for Adverse Events b. XALKORI must be permanently discontinued in case of further Grade ≥ 3 recurrence. See sections 4.4 and 4.8. c. See sections 4.4 and 4.8. d. Heart rate less than 60 beats per minute (bpm). e. Permanently discontinue for recurrence. Hepatic impairment XALKORI has not been studied in patients with hepatic impairment. Clinical studies that were conducted excluded patients with AST or ALT >2.5×upper limit of normal (ULN), or if due to underlying malignancy, >5.0×ULN or with total bilirubin >1.5×ULN. Treatment with XALKORI should be used with caution in patients with mild and moderate hepatic impairment. XALKORI should not be used in patients with severe hepatic impairment (see sections 4.3, 4.4 and 4.8). Renal impairment No starting dose adjustment is recommended for patients with mild (60 ≤ creatinine clearance [CLcr] < 90 mL/min) or moderate (30 ≤ CLcr < 60 mL/min) renal impairment, since the population pharmacokinetic analysis indicated no clinically meaningful changes in steady-state crizotinib exposure in these patients. Crizotinib plasma concentrations may be increased in patients with severe renal impairment (CLcr <30 mL/min). The crizotinib dose should be adjusted to 250 mg taken orally once daily in patients with severe renal impairment not requiring peritoneal dialysis or hemodialysis. The dose may be increased to 200 mg twice daily based on individual safety and tolerability after at least 4 weeks of treatment (see sections 4.4 and 5.2). Elderly Of the 172 crizotinib-treated patients in randomised Phase 3 Study 1, 27 (16%) were 65 years or older. Of the 149 patients in Study A, 20 (13%) were 65 years or older. Of the 934 patients in Study B, 152 (16%) were 65 years or older (see section 5.2). No overall differences in safety or efficacy were observed in comparison with younger patients. Paediatric population The safety and efficacy of XALKORI in paediatric patients has not been established. No data are available. Method of administration The capsules should be swallowed whole preferably with water, and should not be crushed, dissolved, or opened. They may be taken with or without food. Grapefruit or grapefruit juice should be avoided since it may increase crizotinib plasma concentration; St. John's wort should be avoided since it may decrease crizotinib plasma concentration (see section 4.5). 4.3 Contraindications Hypersensitivity to crizotinib or to any of the excipients listed in section 6.1. Severe hepatic impairment (see sections 4.2, 4.4 and 4.8). 4.4 Special warnings and precautions for use Assessment of ALK status When assessing the ALK status of a patient, it is important that a well-validated and robust methodology is chosen to avoid false negative or false positive determinations. Hepatotoxicity Drug-induced hepatotoxicity with fatal outcome has occurred. These cases have occurred during XALKORI treatment in less than 1% of patients in clinical trials. Concurrent elevations in ALT greater than 3×ULN and total bilirubin greater than 2×ULN without elevated alkaline phosphatase have been observed in less than 1% patients in clinical trials. Increases to Grade 3 or 4 ALT elevations were observed in 17% of patients receiving crizotinib versus 4% of patients receiving chemotherapy in randomised Phase 3 Study 1. Increases to Grade 3 or 4 ALT elevations were observed in 7% of patients in Study A and 8% of patients in Study B. Grade 3 and 4 elevations were generally reversible upon dosing interruption. Two patients from randomised Phase 3 Study 1 (1%), 1 patient from Study A (< 1%) and 6 patients from Study B (< 1%) required permanent discontinuation from treatment. Transaminase elevations generally occurred within the first 2 months of treatment. XALKORI should not be used in patients with severe hepatic impairment (including patients with total bilirubin > 3 × ULN regardless of ALT/AST) (see sections 4.2, 4.3 and 4.8). Liver function tests including ALT, AST, and total bilirubin should be monitored once a week during the first 2 months of treatment, then once a month and as clinically indicated, with more frequent repeat testing for Grades 2, 3 or 4 elevations. For patients who develop transaminase elevations, see section 4.2. Interstitial lung disease/Pneumonitis Severe, life-threatening, and/or fatal interstitial lung disease (ILD)/pneumonitis can occur in patients treated with XALKORI. Across randomized Phase 3 Study 1 and Studies A and B (n=1255), 33 (2.6%) patients treated with crizotinib had any grade ILD, including 13 (1%) patients with Grade 3 or 4, and 6 (0.5%) patients with fatal cases. These cases generally occurred within 2 months after the initiation of treatment. Patients with pulmonary symptoms indicative of ILD/pneumonitis should be monitored. XALKORI treatment should be withheld if ILD/pneumonitis is suspected. Drug-induced ILD/pneumonitis should be considered in the differential diagnosis of patients with ILD-like conditions such as: pneumonitis, radiation pneumonitis, hypersensitivity pneumonitis, interstitial pneumonitis, pulmonary fibrosis, acute respiratory distress syndrome (ARDS), alveolitis, lung infiltration, pneumonia, pulmonary edema, chronic obstructive pulmonary disease, pleural effusion, aspiration pneumonia, bronchitis, obliterative bronchiolitis, and bronchiectasis. Other potential causes of ILD/pneumonitis should be excluded, and XALKORI should be permanently discontinued in patients diagnosed with treatment-related ILD/pneumonitis (see sections 4.2 and 4.8). QT interval prolongation QTc prolongation has been observed in clinical studies in patients treated with XALKORI (see sections 4.8 and 5.2) which may lead to an increased risk for ventricular tachyarrhythmias (e.g., Torsade de Pointes) or sudden death. The benefits and potential risks of crizotinib should be considered before beginning therapy in patients with pre-existing bradycardia, who have a history of or predisposition for QTc prolongation, who are taking antiarrhythmics or other medicinal products that are known to prolong QT interval and in patients with relevant pre-existing cardiac disease and/or electrolyte disturbances. XALKORI should be administered with caution in these patients and periodic monitoring of electrocardiograms (ECG), electrolytes and renal function is required. When using XALKORI, ECG and electrolytes (e.g., calcium, magnesium, potassium) should be obtained as close as possible prior to the first dose and periodic monitoring with ECGs and electrolytes is recommended, especially at the beginning of treatment in case of vomiting, diarrhoea, dehydration or impaired renal function. Correct electrolytes as necessary. If QTc increases by greater than or equal to 60 msec from baseline but QTc is < 500 msec, crizotinib should be withheld and cardiologist advice should be sought. If QTc increases to greater than or equal to 500 msec, cardiologist advice must be immediately sought. For patients who develop QTc prolongation, see sections 4.2, 4.8 and 5.2. Bradycardia Treatment-emergent all-causality bradycardia was reported in clinical studies in 5 to 10% of patients treated with crizotinib. Symptomatic bradycardia (e.g., syncope, dizziness, hypotension) can occur in patients receiving XALKORI. The full effect of crizotinib on reduction of heart rate may not develop until several weeks after start of treatment. Avoid using crizotinib in combination with other bradycardic agents (e.g., beta-blockers, non-dihydropyridine calcium channel blockers such as verapamil and diltiazem, clonidine, digoxin) to the extent possible, due to the increased risk of symptomatic bradycardia. Monitor heart rate and blood pressure regularly. Dose modification is not required in cases of asymptomatic bradycardia. For management of patients who develop symptomatic bradycardia, see Dose Modification and Undesirable Effects sections (see sections 4.2 and 4.8). Neutropenia and Leukopenia In clinical trials with crizotinib (randomised Phase 3 Study 1 and Studies A and B) Grade 3 or 4 neutropenia has been very commonly (6%-13%) reported. Grade 3 or 4 leukopenia has been commonly (2%) reported (see section 4.8). Less than 1% of patients experienced febrile neutropenia in clinical trials with crizotinib. Complete blood counts including differential white blood cell counts should be monitored as clinically indicated, with more frequent repeat testing if Grade 3 or 4 abnormalities are observed, or if fever or infection occurs (see section 4.2). Gastrointestinal perforation In clinical studies with crizotinib, events of gastrointestinal perforations were reported. There were reports of fatal cases of gastrointestinal perforation during post-marketing use of XALKORI (see section 4.8). Crizotinib should be used with caution in patients at risk for gastrointestinal perforation (e.g., history of diverticulitis, metastases to the gastrointestinal tract, concomitant use of medications with a recognized risk of gastrointestinal perforation). Crizotinib should be discontinued in patients who develop gastrointestinal perforation. Patients should be informed of the first signs of gastrointestinal perforations and be advised to consult rapidly in case of occurrence. Renal impairment If patients have severe renal impairment not requiring peritoneal dialysis or hemodialysis, the dose of crizotinib should be adjusted (see sections 4.2 and 5.2). Visual effects Vision disorder occurred in patients in randomised Phase 3 Study 1, Study A and Study B. Ophthalmological evaluation (e.g., visual acuity, fundoscopy, and slit lamp examinations) should be considered if vision disorder persists or worsens in severity (see section 4.8). Drug-drug interactions The concomitant use of crizotinib with strong CYP3A4 inhibitors/inducers and CYP3A4 substrates with narrow therapeutic indices should be avoided (see section 4.5). Avoid using crizotinib in combination with other bradycardic agents, medicinal products that are known to prolong QT interval and/or antiarrhythmics (see section 4.4 QT interval prolongation, Bradycardia, and section 4.5). Non-adenocarcinoma histology Limited information is available in patients with ALK-positive NSCLC with non-adenocarcinoma histology. (see section 5.1). 4.5 Interaction with other medicinal products and other forms of interaction Pharmacokinetic interactions Agents that may increase crizotinib plasma concentrations Coadministration of crizotinib with strong CYP3A inhibitors may increase crizotinib plasma concentrations. Coadministration of a single 150 mg oral dose of crizotinib in the presence of ketoconazole (200 mg twice daily), a strong CYP3A inhibitor, resulted in increases in crizotinib systemic exposure, with crizotinib AUCinf and Cmax values that were approximately 3.2-fold and 1.4-fold, respectively, those seen when crizotinib was administered alone. Therefore, the concomitant use of strong CYP3A inhibitors (certain protease inhibitors like atazanavir, indinavir, nelfinavir, ritonavir, saquinavir, and, certain azole antifungals like itraconazole, ketoconazole, and voriconazole, certain macrolides like clarithromycin, telithromycin, and troleandomycin) should be avoided. Grapefruit or grapefruit juice may also increase plasma concentrations of crizotinib and should be avoided (see sections 4.2 and 4.4). Furthermore, the effect of CYP3A inhibitors on steady-state crizotinib exposure has not been established. Agents that may decrease crizotinib plasma concentrations Coadministration of a single 250 mg crizotinib dose with rifampicin (600 mg QD), a strong CYP3A4 inducer, resulted in 82% and 69% decreases in crizotinib AUCinf and Cmax, respectively, compared to when crizotinib was given alone. Coadministration of crizotinib with strong CYP3A inducers may decrease crizotinib plasma concentrations. The concurrent use of strong CYP3A inducers, including but not limited to carbamazepine, phenobarbital, phenytoin, rifabutin, rifampicin, and St. John's wort, should be avoided (see section 4.4). Furthermore, the effect of CYP3A inducers on steady-state crizotinib exposure has not been established. Coadministration with medicinal products that increase gastric pH The aqueous solubility of crizotinib is pH dependent, with low (acidic) pH resulting in higher solubility. Administration of a single 250 mg crizotinib dose following treatment with esomeprazole 40 mg QD for 5 days resulted in an approximately 10% decrease in crizotinib total exposure (AUCinf) and no change in peak exposure (Cmax); the extent of the change in total exposure was not clinically meaningful. Therefore, starting dose adjustment is not required when crizotinib is coadministered with agents that increase gastric pH (such as proton-pump inhibitors, H2 blockers, or antacids). Agents whose plasma concentrations may be altered by crizotinib Following 28 days of crizotinib dosing at 250 mg taken twice daily in cancer patients, the oral midazolam AUC was 3.7-fold of those seen when midazolam was administered alone, suggesting that crizotinib is a moderate inhibitor of CYP3A. Therefore, coadministration of crizotinib with CYP3A substrates with narrow therapeutic indices, including but not limited to alfentanil, cisapride, cyclosporine, ergot derivatives, fentanyl, pimozide, quinidine, sirolimus, and tacrolimus should be avoided (see section 4.4). If the combination is needed, then close clinical monitoring should be exercised. In vitro studies indicated that crizotinib is an inhibitor of CYP2B6. Therefore, crizotinib may have the potential to increase plasma concentrations of coadministered drugs that are metabolized by CYP2B6 (e.g., bupropion, efavirenz). In vitro studies in human hepatocytes indicated that crizotinib may induce pregnane X receptor (PXR)- and constitutive androstane receptor (CAR)-regulated enzymes (e.g., CYP3A4, CYP2B6, CYP2C8, CYP2C9, UGT1A1). However, there was no observed induction in vivo when crizotinib was coadministered with the CYP3A probe substrate midazolam. Caution should be exercised in administering crizotinib in combination with medicinal products that are predominantly metabolised by these enzymes. Of note, the effectiveness of concomitant administration of oral contraceptives may be reduced. In vitro studies indicated that crizotinib is a weak inhibitor of UGT1A1 and UGT2B7. Therefore, crizotinib may have the potential to increase plasma concentrations of coadministered drugs that are metabolized predominantly by UGT1A1 (e.g., raltegravir, irinotecan) or UGT2B7 (e.g., morphine, naloxone). Based on an in vitro study, crizotinib is predicted to inhibit intestinal P-gp. Therefore, administration of crizotinib with medicinal products that are substrates of P-gp (e.g., digoxin, dabigatran, colchicine, pravastatin) may increase their therapeutic effect and adverse reactions. Close clinical surveillance is recommended when crizotinib is administered with these medicinal products. Crizotinib is an inhibitor of OCT1 and OCT2 in vitro. Therefore, crizotinib may have the potential to increase plasma concentrations of coadministered drugs that are substrates of OCT1 or OCT2 (e.g., metformin, procainamide). Pharmacodynamic interactions In clinical studies, prolonged QT interval was observed with crizotinib. Therefore, the concomitant use of crizotinib with medicinal products known to prolong QT interval or medicinal products able to induce Torsades de pointes (e.g., class IA [quinidine, disopyramide] or class III [e.g., amiodarone, sotalol, dofetilide, ibutilide], methadone, cisapride, moxifloxacine, antipsychotics, etc.) should be carefully considered. A monitoring of the QT interval should be made in case of combinations of such medicinal products (see sections 4.2 and 4.4). Bradycardia has been reported during clinical studies; therefore, use crizotinib with caution due to the risk of excessive bradycardia when used in combination with other bradycardic agents (e.g., non-dihydropyridine calcium channel blockers such as verapamil and diltiazem, beta-blockers, clonidine, guanfacine, digoxin, mefloquine, anticholinesterases, pilocarpine) (see sections 4.2 and 4.4). 4.6 Fertility, pregnancy and lactation Contraception in males and females Women of childbearing potential should be advised to avoid becoming pregnant while receiving XALKORI. Adequate contraceptive methods should be used during therapy, and for at least 90 days after completing therapy (see section 4.5). Pregnancy XALKORI may cause foetal harm when administered to a pregnant woman. Studies in animals have shown reproductive toxicity (see section 5.3). There are no data in pregnant women using crizotinib. This medicinal product should not be used during pregnancy unless the clinical condition of the mother requires treatment. Pregnant women, or patients becoming pregnant while receiving crizotinib, or treated male patients as partners of pregnant women, should be apprised of the potential hazard to the foetus. Breast-feeding It is not known whether crizotinib and its metabolites are excreted in human milk. Because of the potential harm to the infant, mothers should be advised to avoid breast-feeding while receiving XALKORI (see section 5.3). Fertility Based on nonclinical safety findings, male and female fertility may be compromised by treatment with XALKORI (see section 5.3). Both men and women should seek advice on fertility preservation before treatment. 4.7 Effects on ability to drive and use machines Caution should be exercised when driving or operating machines as patients may experience symptomatic bradycardia (e.g., syncope, dizziness, hypotension), vision disorder, or fatigue while taking XALKORI (see sections 4.2, 4.4 and 4.8). 4.8 Undesirable effects Summary of the safety profile Data described below reflect exposure to XALKORI in 172 patients with ALK-positive advanced NSCLC who participated in randomised Phase 3 Study 1 and in 1083 patients with ALK-positive advanced NSCLC who participated in 2 single-arm clinical trials (Studies A and B). These patients received a starting oral dose of 250 mg taken twice daily continuously. The most serious adverse reactions in patients with ALK-positive advanced NSCLC are hepatotoxicity, ILD/pneumonitis, neutropenia and QT interval prolongation (see section 4.4). The most common adverse reactions (≥25%) in patients with ALK-positive NSCLC are vision disorder, nausea, diarrhoea, vomiting, constipation, oedema, elevated transaminases, and fatigue. Tabulated list of adverse reactions Table 3 presents adverse reactions reported in patients with ALK-positive advanced NSCLC who received crizotinib in randomised Phase 3 Study 1 with median duration of study treatment 31 weeks. The most frequent adverse reactions that led to dosing interruptions were neutropenia (8 %), elevated transaminases (8%), nausea (5%) and vomiting (3%). The most frequent adverse reactions that led to dose reductions were elevated transaminases (8%), electrocardiogram QT prolonged (3%), and neutropenia (2%). Investigator-assessed treatment-related adverse events resulting in permanent discontinuation occurred in 11 (6%) patients on crizotinib. The most frequent adverse reactions that led to permanent discontinuation were interstitial lung disease (2%) and elevated transaminases (1%). The adverse reactions listed in Table 3 are presented by system organ class and frequency categories are defined using the following convention: very common (greater than or equal to 1/10); common (greater than or equal to 1/100 to less than 1/10), uncommon (greater than or equal to 1/1,000 to less than 1/100) or rare (greater than or equal to 1/10,000 to less than 1/1,000). Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. Table 3. Adverse reactions reported in crizotinib randomised Phase 3 Study 1
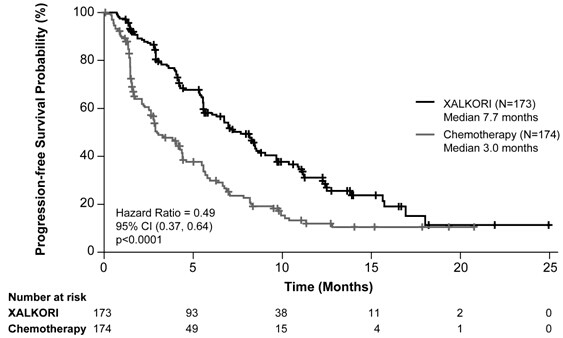
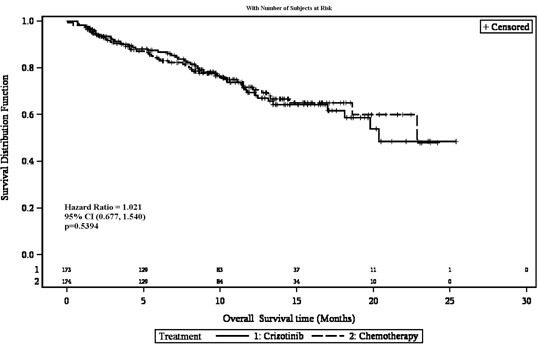
Includes cases reported within the clustered terms: a. Neutropenia (Febrile neutropenia, Neutropenia, Neutrophil count decreased) b. Anaemia (Anaemia, Haemoglobin decreased) c. Neuropathy (Dysaesthesia, Gait disturbance, Hypoaesthesia, Muscular weakness, Neuralgia, Neuropathy peripheral, Paraesthesia, Peripheral sensory neuropathy, Polyneuropathy, Skin burning sensation) d. Dizziness (Balance disorder, Dizziness, Dizziness postural) e. Vision disorder (Diplopia, Photophobia, Photopsia, Vision blurred, Visual acuity reduced, Visual impairment, Vitreous floaters) f. Bradycardia (Bradycardia, Sinus bradycardia) g. Interstitial Lung Disease (Acute respiratory distress syndrome, Interstitial lung disease, Pneumonitis) h. Elevated Transaminases (Alanine aminotransferase increased, Aspartate aminotransferase increased, Gamma-glutamyltransferase increased, Hepatic function abnormal, Transaminases increased) i. Renal Cyst (Renal cyst) j. Oedema (Face oedema, Generalised oedema, Local swelling, Localised oedema, Oedema, Oedema peripheral, Periorbital oedema) The safety analysis population in Study B included 934 patients who received crizotinib. The median duration of treatment was 23 weeks. The most common adverse reactions (≥25%) included vision disorder, nausea, vomiting, diarrhoea, oedema, constipation, and fatigue. The most common Grade 3 or 4 adverse reactions (>3%) in Study B were neutropenia, elevated transaminases, and constipation. The safety analysis population in Study A included 149 patients who received crizotinib. The median duration of treatment was 43 weeks. The most common adverse reactions (≥25%) in Study A included nausea, vision disorder, diarrhoea, vomiting, oedema, constipation, dizziness, fatigue and decreased appetite. Description of selected adverse reactions Hepatotoxicity Drug-induced hepatotoxicity with fatal outcome has occurred. These cases have occurred during XALKORI treatment in less than 1% of patients in clinical trials. Concurrent elevations in ALT greater than 3×ULN and total bilirubin greater than 2×ULN without elevated alkaline phosphatase have been observed in less than 1% patients in clinical trials. Increases to Grade 3 or 4 ALT elevation were observed in 17% of patients receiving crizotinib versus 4% of patients receiving chemotherapy in randomised Phase 3 Study 1. Increases to Grade 3 or 4 ALT elevations were observed in 7% of patients in Study A and 8% of patients in Study B. Transaminase (ALT, AST) elevations generally occurred within the first 2 months of treatment. Across Study A, Study B and randomised Phase 3 Study 1, median times to onset of increased Grade 1 or 2 transaminases ranged from 22 to 26 days. Median times to onset of increased Grade 3 or 4 transaminases ranged from 30 to 43 days. Grade 3 and 4 elevations were generally reversible upon dosing interruption. Dose reductions associated with transaminase elevations occurred in 8% of patients from randomised Phase 3 Study 1 and 3% of patients from Study A and Study B. Two patients from randomised Phase 3 Study 1 (1%), 1 patient from Study A (<1%) and 6 patients from Study B (< 1%) required permanent discontinuation from treatment. XALKORI should not be used in patients with severe hepatic impairment (see sections 4.2, 4.3 and 4.4). Liver function tests including ALT, AST, and total bilirubin should be monitored once a week during the first 2 months of treatment, then once a month and as clinically indicated, with more frequent repeat testing for Grades 2, 3 or 4 elevation. Patients should be monitored for hepatotoxicity and managed as recommended in sections 4.2 and 4.4. Gastrointestinal effects Nausea, diarrhoea, vomiting, and constipation were the most commonly reported gastrointestinal events. Median times to onset for nausea and vomiting were 2 to 3 days. Most events of these were mild to moderate in severity, and declined in frequency after 3 to 4 weeks. Supportive care should include the use of antiemetic medicinal products. Diarrhoea and constipation were primarily mild to moderate in severity. Supportive care for diarrhoea and constipation should include the use of standard antidiarrheal and laxative medicinal products, respectively. In clinical studies with crizotinib, events of gastrointestinal perforations were reported. There were reports of fatal cases of gastrointestinal perforation during post-marketing use of XALKORI (see section 4.4). QT interval prolongation The mean changes from baseline for QTcF (corrected QT by the Fridericia method) on Cycle 1 Day 1 and Cycle 2 Day 1 were 8.3 msec and 8.7 msec in randomised Phase 3 Study 1 and study B, respectively (highest upper bound of 2-sided 90% CI for QTcF were 13.1 msec and 10.2 msec respectively and was <15 msec, which is unlikely to constitute a clinically relevant QTc effect). A QTcF ≥500 msec was recorded in 5 (3.4%) and 10 (1.1%) patients in randomised Phase 3 Study 1 and B, respectively and a maximum increase from baseline in QTcF ≥60 msec was observed in 9 (6.3%) and 38 (4.3%) patients in randomised Phase 3 Study 1 and B, respectively. In randomised Phase 3 Study 1, all-causality Grade 3 or 4 Electrocardiogram QT prolonged was reported in 6 (3.5%) patients (see sections 4.2, 4.4, 4.5 and 5.2). QT prolongation can result in arrhythmias and is a risk factor for sudden death. QT prolongation may clinically manifest as bradycardia, dizziness, and syncope. Electrolyte disturbances, dehydration and bradycardia may further increase the risk of QTc prolongation and thus, periodic monitoring of ECG and electrolyte levels is recommended in patients with GI toxicity (see section 4.4). Bradycardia Treatment-emergent all-causality bradycardia was reported by 8 (5%) patients in randomised Phase 3 Study 1 and 13 (9%) and 108 (10%) patients in Studies A and B, respectively. In Studies 1, A, and B, 19 of 170 (11%) patients, 26 of 144 (18%) patients, and 90 of 890 (10%) patients, respectively, had a heart rate <50 bpm. The use of concomitant medications associated with bradycardia should be carefully evaluated. Patients who develop symptomatic bradycardia should be managed as recommended in the Dose Modification and Warnings and Precautions sections (see sections 4.2, 4.4 and 4.5). Interstitial lung disease/Pneumonitis Severe, life-threatening, and/or fatal interstitial lung disease (ILD)/pneumonitis can occur in patients treated with XALKORI. Across randomized Phase 3 Study 1 and Studies A and B (n=1255), 33 (2.6%) patients treated with crizotinib had any grade ILD, 13 (1%) patients had Grade 3 or 4, and 6 (0.5%) patients had fatal outcome. These cases generally occurred within 2 months after the initiation of treatment. Patients with pulmonary symptoms indicative of ILD/pneumonitis should be monitored. Other potential causes of ILD/pneumonitis should be excluded, (see sections 4.2 and 4.4). Visual effects Treatment-emergent all-causality vision disorder, most commonly visual impairment, photopsia, vision blurred, and vitreous floaters was experienced by 103 (60%) patients in randomised Phase 3 Study 1 and in 99 (66%) and 513 (55%) patients in Study A and Study B, respectively. This event was reported as mild (96%), moderate (3%), and severe (< 1%) with median times to onset of 5 days, 15 days and 7 days in Studies 1, A, and B, respectively. No patients from randomised Phase 3 Study 1, 1 patient from Study A and 4 patients from Study B had temporary treatment discontinuation. One patient from randomised Phase 3 Study 1 and 1 patient from Study B had a dose reduction for vision disorder. Permanent discontinuation from crizotinib treatment due to vision disorder was not required for any patients in Studies 1, A, or B. Ophthalmological evaluation should be considered if vision disorder persists or worsens in severity (see section 4.4). Based on the Visual Symptom Assessment Questionnaire (VSAQ-ALK), patients treated with XALKORI in randomised Phase 3 Study 1 reported a higher incidence of visual disturbances compared to patients treated with chemotherapy. The onset of vision disorders generally started within the first week of drug administration. The majority (of patients on the XALKORI arm in randomised Phase 3 Study 1 (> 50%) reported visual disturbances; which occurred at a frequency of 4 to 7 days each week, lasted up to 1 minute, and had mild or no impact (scores 0 to 3 out of a maximum score of 10) on daily activities as captured in a patient questionnaire. Nervous system effects Treatment-emergent all-causality neuropathy as defined in Table 3, was experienced by 33 (19%) patients in randomised Phase 3 Study 1 and 36 (24%) and 178 (19%) patients in Studies A and B, respectively. Dysgeusia was also very commonly reported in these studies, and was primarily Grade 1 in severity. Renal cyst Treatment-emergent all-causality complex renal cysts were experienced by 7 (4%) patients in randomised Phase 3 Study 1 and 1 (< 1%) and 12 (1%) patients in Studies A and B, respectively. Local cystic invasion beyond the kidney was observed in some patients. Periodic monitoring with imaging and urinalysis should be considered in patients who develop renal cysts. Neutropenia and Leukopenia Grade 3 or 4 neutropenia was observed in 13%, 6% and 11% of patients treated with crizotinib in randomised Phase 3 Study 1, Study A and Study B, respectively. Median times to onset of any Grade neutropenia was 43, 197 and 47 days, respectively; median times to onset of Grade 3 or 4 neutropenia was 165, 197 and 64 days, respectively. Neutropenia was associated with dose reduction for 2%, 1% and 3% of crizotinib-treated patients in randomised Phase 3 Study 1, Study A and Study B, respectively. Neutropenia was associated with permanent treatment discontinuation for <1% of patients in Study B. There were no permanent treatment discontinuation due to neutropenia in Study A and randomised Phase 3 Study 1. Less than 1% of patients experienced febrile neutropenia in clinical trials with crizotinib. Grade 3 or Grade 4 leukopenia was observed in 2% of patients in both the randomised Phase 3 Study 1 and Study B and in <1% of patients in Study A. In randomised Phase 3 Study 1, Study A and Study B, median times to onset of any Grade leukopenia was 64, 75 and 43 days, respectively; median times to onset of Grade 3 or 4 leukopenia was 373, 299 and 75 days, respectively. Leukopenia was associated with a dose reduction for <1% of patients in randomised Phase 3 Study 1 and Study B. There were no dose reductions associated with leukopenia in Study A. Leukopenia was not associated with permanent treatment discontinuation for any patients in randomised Phase 3 Study 1, Study A or Study B. In randomised Phase 3 Study 1, shifts to Grade 3 or 4 decreases in leukocytes and neutrophils were observed at frequencies of 5% and 13%, respectively. In Study A, shifts to Grade 3 or 4 decreases in leukocytes and neutrophils were observed at frequencies of <3% and 8%, respectively. In Study B, shifts to Grade 3 or 4 decreases in leukocytes and neutrophils were observed at a frequency of <3% and 8%, respectively. Complete blood counts including differential white blood cell counts should be monitored as clinically indicated, with more frequent repeat testing if Grade 3 or 4 abnormalities are observed, or if fever or infection occurs. For patients who develop hematologic laboratory abnormalities, see section 4.2. Reporting of suspected adverse reactions Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the internet at www.mhra.gov.uk/yellowcard. 4.9 Overdose Treatment of overdose with the medicinal product consists of general supportive measures. There is no antidote for XALKORI. 5. Pharmacological properties 5.1 Pharmacodynamic properties Pharmacotherapeutic group: Anti-neoplastic agents, protein kinase inhibitor; ATC code: L01XE16. Mechanism of action Crizotinib is a selective small-molecule inhibitor of the ALK receptor tyrosine kinase (RTK) and its oncogenic variants (i.e. ALK fusion events and selected ALK mutations). Crizotinib is also an inhibitor of the Hepatocyte Growth Factor Receptor (HGFR, c-Met) RTK, and Recepteur d'Origine Nantais (RON) RTK. Crizotinib demonstrated concentration-dependent inhibition of the kinase activity of ALK and c-Met in biochemical assays and inhibited phosphorylation and modulated kinase-dependent phenotypes in cell-based assays. Crizotinib demonstrated potent and selective growth inhibitory activity and induced apoptosis in tumour cell lines exhibiting ALK fusion events (including EML4-ALK and NPM-ALK), or exhibiting amplification of the ALK or MET gene locus. Crizotinib demonstrated anti-tumour efficacy, including marked cytoreductive anti-tumour activity, in mice bearing tumour xenografts that expressed ALK fusion proteins. The anti-tumour efficacy of crizotinib was dose-dependent and correlated to pharmacodynamic inhibition of phosphorylation of ALK fusion proteins (including EML4-ALK and NPM-ALK) in tumours in vivo. Clinical studies Randomised Phase 3 Study 1 The use of single-agent crizotinib in the treatment of ALK-positive advanced NSCLC with or without brain metastases was investigated in 2 a multicenter, multinational, randomised, open-label Phase 3 study (Study 1). The primary objective of this study was to demonstrate that crizotinib 250 mg orally twice daily was superior to second-line standard-of-care chemotherapy (pemetrexed 500 mg/m2 or docetaxel 75 mg/m2) intravenously (IV) every 21 days in prolonging Progression-Free Survival (PFS) in patients with ALK-positive advanced NSCLC who were to receive only 1 prior platinum-based chemotherapy regimen for advanced NSCLC. Patients were required to have ALK-positive NSCLC identified by the Vysis ALK Break Apart FISH Probe Kit from the central laboratory prior to randomization. For patients randomized to the chemotherapy arm, the first choice was to be pemetrexed unless patients received pemetrexed as part of their prior therapy or had squamous histology. The primary efficacy endpoint was PFS with disease progression events determined by IRR. Secondary endpoints included Objective Response Rate (ORR) as determined by independent radiology review (IRR), Duration of Response (DR), Overall Survival (OS), 6-month and 1-year OS probabilities, and Patient-Reported Outcomes (PRO). Patients could continue treatment as assigned beyond the time of RECIST-defined disease progression, as assessed by IRR, at the discretion of the investigator if the patient was perceived to be experiencing clinical benefit. Patients randomised to chemotherapy could cross over to receive crizotinib upon RECIST-defined disease progression confirmed by IRR. The full analysis population for Study 1 included 347 patients with ALK-positive advanced NSCLC. One hundred seventy-three (173) patients were randomised to the crizotinib arm (172 patients received crizotinib) and 174 patients were randomised to the chemotherapy arm (99 [58%] patients received pemetrexed and 72 [42%] patients received docetaxel). Randomization was stratified by ECOG performance status (0-1, 2), brain metastases (present, absent), and prior EGFR tyrosine kinase inhibitor treatment (yes, no). The median duration of study treatment was 31 weeks in the crizotinib arm as compared to 12 weeks in the chemotherapy arm. Fifty eight of 84 (69%) crizotinib-treated patients and 17 of 119 (14%) chemotherapy-treated patients continued treatment for at least 3 weeks after objective disease progression. Key demographic and baseline characteristics for patients in this study were comparable between the crizotinib and chemotherapy arms. The demographic characteristics of the overall study population were 56% female, median age of 50 years, baseline ECOG performance status 0 (39%) or 1 (52%), 52% White and 45% Asian, 4% current smokers, 33% past-smokers, and 63% never smokers. The disease characteristics were metastatic disease in 93% of patients and in 93% of patients' tumors were classified as adenocarcinoma histology. Crizotinib significantly prolonged PFS compared to chemotherapy as assessed by IRR (see Table 4 and Figure 1). The PFS benefit of crizotinib was consistent across subgroups of baseline patient characteristics such as age, gender, race, smoking class, time since diagnosis, ECOG performance status score, presence of brain metastases and prior EGFR TKI therapy. Crizotinib also significantly improved IRR-assessed ORR as compared to chemotherapy. Median DR was 32.1 weeks (95% CI: 26.4, 42.3) in the crizotinib arm and 24.4 weeks (95% CI: 15.0, 36.0) in the chemotherapy arm. Overall survival (OS) data were not mature at the time of the PFS analysis. There was no statistically significant difference between crizotinib and chemotherapy in the preliminary OS analysis that was not adjusted for the potentially confounding effects of crossover. Of the 174 patients in the chemotherapy arm, 112 (64.4%) patients subsequently received crizotinib treatment. Efficacy data from randomised Phase 3 Study 1 are summarized in Table 4, and the Kaplan-Meier curve for PFS is shown in Figure 1. The Kaplan-Meier curve for OS is shown in Figure 2. Table 4. ALK-positive advanced NSCLC efficacy results from randomised phase 3 Study 1 (full analysis population)
HR = Hazard Ratio; CI = confidence interval; NR= not reached a. The median PFS was 4.2 months (95% CI: 2.8, 5.7) with pemetrexed (HR=0.59; p=0.0004 for XALKORI compared with pemetrexed) and 2.6 months (95% CI: 1.6, 4.0) with docetaxel (HR=0.30; p<0.0001 for XALKORI compared with docetaxel). b. Based on the Cox proportional hazards stratified analysis. c. Based on the stratified Log-rank test. d. Interim OS analysis conducted at 40% of total events required for final analysis. e. Estimated using the Kaplan-Meier method. f. The ORR was 29% (95% CI: 21%, 39%) with pemetrexed (p<0.0001 compared with XALKORI) and 7% (95% CI: 2%, 16%) with docetaxel (p<0.0001 compared with XALKORI). g. Based on the stratified Cochran-Mantel-Haenszel test. Figure 1. Kaplan-Meier curves for progression-free survival (based on IRR) by treatment arm in randomised phase 3 Study 1 (full analysis population)
Figure 2. Kaplan-Meier curves for overall survival by treatment arm in randomized phase 3 Study 1 (full analysis population)
CI = confidence interval | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||