|
英文药名:Halaven(eribulin mesylate) 中文药名:甲磺酸艾日布林注射液 生产厂家:卫材(Eisa)
Halaven 0.44 mg/ml solution for injection
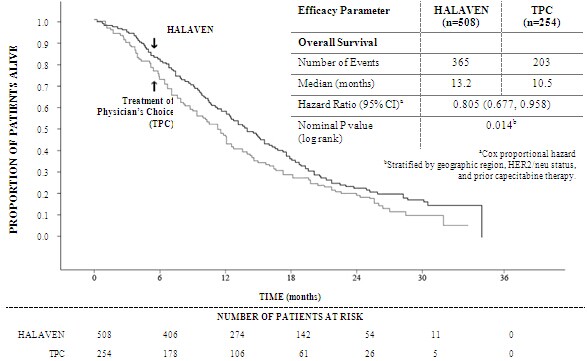
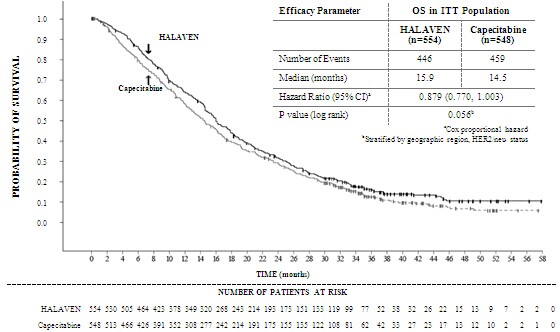
Patients with hepatic impairment Impaired liver function due to metastases: The recommended dose of eribulin in patients with mild hepatic impairment (Child-Pugh A) is 0.97 mg/m2 administered intravenously over 2 to 5 minutes on Days 1 and 8 of a 21-day cycle. The recommended dose of eribulin in patients with moderate hepatic impairment (Child-Pugh B) is 0.62 mg/m2 administered intravenously over 2 to 5 minutes on Days 1 and 8 of a 21-day cycle. Severe hepatic impairment (Child-Pugh C) has not been studied but it is expected that a more marked dose reduction is needed if eribulin is used in these patients. Impaired liver function due to cirrhosis: This patient group has not been studied. The doses above may be used in mild and moderate impairment but close monitoring is advised as the doses may need readjustment. Patients with renal impairment Patients with severely impaired renal function (creatinine clearance <40 ml/min) may need a reduction of the dose. The optimal dose for this patient groups remains to be established. Caution and close safety monitoring as advised. No specific dose adjustments are recommended for patients with mild to moderate renal impairment but close safety monitoring is advised. (See section 5.2) Elderly patients No specific dose adjustments are recommended based on the age of the patient (see section 4.8). Paediatric population There is no relevant use of HALAVEN in children and adolescents in the indication of breast cancer. Method of administration The dose may be diluted in up to 100 ml of sodium chloride 9 mg/ml (0.9%) solution for injection. It should not be diluted in glucose 5% infusion solution. For instructions on the dilution of the medicinal product before administration, see section 6.6. Good peripheral venous access, or a patent central line, should be ensured prior to administration. There is no evidence that eribulin mesilate is a vesicant or an irritant. In the event of extravasation, treatment should be symptomatic. For information relevant to the handling of cytotoxic drugs see section 6.6. 4.3 Contraindications - Hypersensitivity to the active substance or to any of the excipients listed in section 6.1 - Breast-feeding 4.4 Special warnings and precautions for use Haematology Myelosuppression is dose dependent and primarily manifested as neutropenia (section 4.8). Monitoring of complete blood counts should be performed on all patients prior to each dose of eribulin. Treatment with eribulin should only be initiated in patients with ANC values ≥ 1.5 x 109/l and platelets > 100 x 109/l. Febrile neutropenia occurred in < 5% of breast cancer patients treated with eribulin. Patients experiencing febrile neutropenia, severe neutropenia or thrombocytopenia, should be treated according to the recommendations in section 4.2. Patients with ALT or AST >3 x ULN experienced a higher incidence of Grade 4 neutropenia and febrile neutropenia. Although data are limited, patients with bilirubin >1.5 x ULN also have a higher incidence of Grade 4 neutropenia and febrile neutropenia. Severe neutropenia may be managed by the use of G-CSF or equivalent at the physician's discretion in accordance with relevant guidelines (see section 5.1). Peripheral neuropathy Patients should be closely monitored for signs of peripheral motor and sensory neuropathy. The development of severe peripheral neurotoxicity requires a delay or reduction of dose (see section 4.2) In clinical trials, patients with pre-existing neuropathy greater than Grade 2 were excluded. However, patients with pre-existing neuropathy Grade 1 or 2 were no more likely to develop new or worsening symptoms than those who entered the study without the condition. QT prolongation In an uncontrolled open-label ECG study in 26 patients, QT prolongation was observed on Day 8, independent of eribulin concentration, with no QT prolongation observed on Day 1. ECG monitoring is recommended if therapy is initiated in patients with congestive heart failure, bradyarrhythmias or concomitant treatment with medicinal products known to prolong the QT interval, including Class Ia and III antiarrhythmics, and electrolyte abnormalities. Hypokalemia or hypomagnesemia should be corrected prior to initiating HALAVEN and these electrolytes should be monitored periodically during therapy. Eribulin should be avoided in patients with congenital long QT syndrome. Use in combination with anti-HER2 therapy There is no experience of using eribulin in combination with anti-HER2 therapy in clinical trials. Excipients This medicinal product contains small amounts of ethanol (alcohol), less than 100 mg per dose. 4.5 Interaction with other medicinal products and other forms of interaction Eribulin is mainly (up to 70%) eliminated through biliary excretion. The transport protein involved in this process is unknown. Complete inhibition of the transport could in theory give rise to a more than 3-fold increase in plasma concentrations. It is not recommended to use substances which are inhibitors of hepatic transport proteins such as organic anion-transporting proteins (OATPs) and multidrug resistant proteins (MRPs) etc concomitantly with eribulin. Inhibitors of such transporters include but are not limited to: cyclosporine, ritonavir, saquinavir, lopinavir and certain other protease inhibitors, efavirenz, emtricitabine, quinine, quinidine, disopyramide etc. It cannot be excluded that concomitant treatment with inducing substances, such as carbamazepine, phenytoin, St John´s wort (Hypericum perforatum), could give rise to reduced plasma concentrations of eribulin, and co-administration with inducers should be carried out with caution considering a potential risk for reduced drug efficacy. No marked effects on eribulin exposure (AUC and Cmax) were observed during treatment with the CYP3A4 inducer rifampicin. However, rifampicin may due to its transporter inhibitory property counteract its possible inducing effect on eribulin elimination. Therefore, the effect of rifampicin may not presently be extrapolated to other inducers. No drug-drug interactions are expected with CYP3A4 inhibitors. Eribulin exposure (AUC and Cmax) was unaffected by ketoconazole, a CYP3A4 and P glycoprotein (Pgp) inhibitor. Effects of eribulin on the pharmacokinetics of other drugs In vitro data indicate that eribulin is a mild inhibitor of the important drug metabolising enzyme CYP3A4. No in vivo data are available. Caution and monitoring for adverse effects is recommended with concomitant use of substances that have a narrow therapeutic window and that are eliminated mainly via CYP3A4-mediated metabolism (e.g. alfentanil, cyclosporine, ergotamine, fentanyl, pimozide, quinidine, sirolimus, tacrolimus). Eribulin does not inhibit the CYP enzymes CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 or 2E1 at relevant clinical concentrations. 4.6 Fertility, pregnancy and lactation Pregnancy There are no data from the use of eribulin in pregnant women. Eribulin is embryotoxic, foetotoxic, and teratogenic in rats. HALAVEN should not be used during pregnancy unless clearly necessary and after a careful consideration of the needs of the mother and the risk to the foetus. Women of childbearing potential must be advised to avoid becoming pregnant whilst they or their male partner are receiving HALAVEN and have to use effective contraception during and up to 3 months after treatment. Breast-feeding It is unknown whether eribulin/metabolites are excreted in human or animal breast milk. A risk to newborns/infants cannot be excluded and therefore HALAVEN must not be used during breast-feeding (see section 4.3). Fertility Testicular toxicity has been observed in rats and dogs (see section 5.3). Male patients should seek advice on conservation of sperm prior to treatment because of the possibility of irreversible infertility due to therapy with HALAVEN. 4.7 Effects on ability to drive and use machines HALAVEN may cause adverse reactions such as tiredness and dizziness which may lead to a minor or moderate influence on the ability to drive or use machines. Patients should be advised not to drive or use machines if they feel tired or dizzy. 4.8 Undesirable effects Summary of safety profile The most commonly reported undesirable effects related to HALAVEN, are bone marrow suppression manifested as neutropenia, leucopenia, anaemia, thrombocytopenia with associated infections. New onset or worsening of pre-existing peripheral neuropathy has also been reported. Gastrointestinal toxicities, manifested as anorexia, nausea, vomiting, diarrhoea, constipation, and stomatitis are among reported undesirable effects. Other undesirable effects include fatigue, alopecia, increased liver enzymes, sepsis and musculoskeletal pain syndrome. Tabulated list of adverse reactions Unless otherwise noted, the table shows the incidence rates of adverse events observed in 1503 breast cancer patients who received the recommended dose in five Phase 2 and two Phase 3 studies. Frequency categories are defined as: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000) and very rare (< 1/10,000). Within each frequency grouping, undesirable effects are presented in order of decreasing frequency. Where Grade 3 or 4 reactions occurred, the actual total frequency and the frequency of Grade 3 or 4 reactions are given. a Includes 1 or 2 Grade 5 b From spontaneous reporting c Includes preferred terms of peripheral neuropathy, peripheral motor neuropathy, polyneuropathy, paraesthesia, peripheral sensory neuropathy, peripheral sensorimotor neuropathy and demyelinating polyneuropathy d No Grade 4 Selected adverse reactions Neutropenia The neutropenia observed was reversible and not cumulative; the mean time to nadir was 13 days and the mean time to recovery from severe neutropenia (< 0.5 x 109/l) was 8 days. Neutrophil counts of < 0.5 x 109/l that lasted for more than 7 days occurred in 13% of breast cancer patients treated with eribulin in the EMBRACE study. Severe neutropenia may be managed by the use of G-CSF or equivalent at the physician's discretion in accordance with relevant guidelines. 18% of breast cancer patients treated in a phase 3 study with eribulin received G-CSF. Neutropenia resulted in discontinuation in < 1% of patients receiving eribulin. Disseminated intravascular coagulation Cases of disseminated intravascular coagulation have been reported, typically in association with neutropenia and/or sepsis. Peripheral neuropathy In the 1503 breast cancer patients the most common adverse reaction resulting in discontinuation of treatment with eribulin was peripheral neuropathy ( 3.3%). The median time to Grade 2 peripheral neuropathy was 85.5 days (post 4 cycles). Development of Grade 3 or 4 peripheral neuropathy occurred in 7.7% of eribulin treated breast cancer patients. In clinical trials, patients with pre-existing neuropathy were as likely to develop new or worsening symptoms as those who entered the study without the condition. In patients with pre-existing Grade 1 or 2 peripheral neuropathy the frequency of treatment-emergent Grade 3 peripheral neuropathy was 14%. Hepatoxicity In some patients with normal/abnormal liver enzymes prior treatment with eribulin, increased levels of liver enzymes have been reported with initiation of eribulin treatment. Such elevations appeared to have occurred early with eribulin treatment in cycle 1 – 2 for the majority of these patients and whilst thought likely to be a phenomenon of adaptation to eribulin treatment by the liver and not a sign of significant liver toxicity in most patients , hepatotoxicity has also been reported. Special populations Elderly population Of the 1503 breast cancer patients treated with the recommended dose of eribulin, 209 patients (13.9%) were > 65 - 75 years of age and 24 patients (1.6%) were > 75 years of age. The safety profile of eribulin in elderly patients (> 65 years of age) was similar to that of patients ≤ 65 years of age except for asthenia/fatigue which showed an increasing trend with age. No dose adjustments are recommended for the elderly population. Patients with hepatic impairment Patients with ALT or AST > 3 x ULN experienced a higher incidence of Grade 4 neutropenia and febrile neutropenia. Although data are limited, patients with bilirubin > 1.5 x ULN also have a higher incidence of Grade 4 neutropenia and febrile neutropenia (see also sections 4.2 and 5.2). Reporting of suspected adverse reactions Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Yellow Card Scheme Website: www.mhra.gov.uk/yellowcard 4.9 Overdose In one case of overdose the patient inadvertently received 7.6 mg of eribulin (approximately 4 times the planned dose) and subsequently developed a hypersensitivity reaction (Grade 3) on Day 3 and neutropenia (Grade 3) on Day 7. Both adverse reactions resolved with supportive care. There is no known antidote for eribulin overdose. In the event of an overdose, the patient should be closely monitored. Management of overdose should include supportive medical interventions to treat the presenting clinical manifestations. 5. Pharmacological properties 5.1 Pharmacodynamic properties Pharmacotherapeutic group: Other antineoplastic agents, ATC code: L01XX41 HALAVEN (eribulin mesilate) is a non-taxane, microtubule dynamics inhibitor belonging to the halichondrin class of antineoplastic agents. It is a structurally simplified synthetic analogue of halichondrin B, a natural product isolated from the marine sponge Halichondria okadai. Eribulin inhibits the growth phase of microtubules without affecting the shortening phase and sequesters tubulin into non-productive aggregates. Eribulin exerts its effects via a tubulin-based antimitotic mechanism leading to G2/M cell-cycle block, disruption of mitotic spindles, and, ultimately, apoptotic cell death after prolonged mitotic blockage. Clinical efficacy The efficacy of HALAVEN in breast cancer is primarily supported by two randomized Phase 3 comparative studies. The 762 patients in the pivotal Phase 3 EMBRACE study (Study 305) had locally recurrent or metastatic breast cancer, and had previously received at least two and a maximum of five chemotherapy regimens, including an anthracycline and a taxane (unless contraindicated). Patients must have progressed within 6 months of their last chemotherapeutic regimen. The HER2 status of the patients was: 16.1% positive, 74.2% negative and 9.7% unknown, whilst 18.9% of patients were triple negative. They were randomized 2:1 to receive either HALAVEN, or treatment of physician's choice (TPC), which consisted of 97% chemotherapy (26% vinorelbine, 18% gemcitabine, 18% capecitabine, 16% taxane, 9% anthracycline, 10% other chemotherapy), or 3% hormonal therapy. The study met its primary endpoint with an overall survival result that was statistically significantly better in the eribulin group compared to TPC at 55% of events. This result was confirmed with an updated overall survival analysis carried out at 77% of events. Study 305 - Updated Overall Survival ( ITT Population)
By independent review, the median progression free survival (PFS) was 3.7 months for eribulin compared to 2.2 months for the TPC arm (HR 0.865, 95% CI: 0.714, 1.048, p=0.137). In response evaluable patients, the objective response rate by the RECIST criteria was 12.2% (95% CI: 9.4%, 15.5%) by independent review for the eribulin arm compared to 4.7% (95% CI: 2.3%, 8.4%) for the TPC arm.
The European Medicines Agency has waived the obligation to submit the results of studies with eribulin in all subsets of the paediatric population in the indication of breast cancer. 5.2 Pharmacokinetic properties Distribution The pharmacokinetics of eribulin are characterized by a rapid distribution phase followed by a prolonged elimination phase, with a mean terminal half-life of approximately 40 h. It has a large volume of distribution (range of means 43 to 114 l/m2). Eribulin is weakly bound to plasma proteins. The plasma protein binding of eribulin (100-1000 ng/ml) ranged from 49% to 65% in human plasma. Biotransformation Unchanged eribulin was the major circulating species in plasma following administration of 14C-eribulin to patients. Metabolite concentrations represented <0.6% of parent compound, confirming that there are no major human metabolites of eribulin. Elimination Eribulin has a low clearance (range of means 1.16 to 2.42 l/h/m2). No significant accumulation of eribulin is observed on weekly administration. The pharmacokinetic properties are not dose or time dependent in the range of eribulin doses of 0.22 to 3.53 mg/m2. Eribulin is eliminated primarily by biliary excretion. The transport protein involved in the excretion is presently unknown. Preclinical in vitro studies indicate that eribulin is transported by Pgp. However it has been shown that at clinically relevant concentrations eribulin is not a Pgp inhibitor in vitro. Additionally, in vivo, concomitant administration of ketoconazole, a Pgp inhibitor, has no effect on eribulin exposure (AUC and Cmax). In vitro studies have also indicated that eribulin is not a substrate for OCT1. After administration of 14C-eribulin to patients, approximately 82% of the dose was eliminated in faeces and 9% in urine indicating that renal clearance is not a significant route of eribulin elimination. Unchanged eribulin represented most of the total radioactivity in faeces and urine. Hepatic impairment A study evaluated the PK of eribulin in patients with mild (Child-Pugh A; n=7) and moderate (Child-Pugh B; n=4) hepatic impairment due to liver metastases. Compared to patients with normal hepatic function (n=6), eribulin exposure increased 1.8-fold and 3-fold in patients with mild and moderate hepatic impairment, respectively. Administration of HALAVEN at a dose of 0.97 mg/m2 to patients with mild hepatic impairment and 0.62 mg/m2 to patients with moderate hepatic impairment resulted in a somewhat higher exposure than after a dose of 1.23 mg/m2 to patients with normal hepatic function. HALAVEN was not studied in patients with severe hepatic impairment (Child-Pugh C). There is no study in patients with hepatic impairment due to cirrhosis. See section 4.2 for dosage recommendation. Renal impairment Data in patients with different degrees of impaired renal function showed that the exposure of eribulin in patients with mild to moderate renal impairment (creatinine clearance ≥ 40 to 80 ml/min) was increased in some patients, as compared to patients with normal renal function. The mean exposure in patients with severe impairment was increased by 75% (creatinine clearance < 40 ml/min, n=4). See section 4.2 for treatment recommendations. 5.3 Preclinical safety data Eribulin was not mutagenic in vitro in the bacterial reverse mutation assay (Ames test). Eribulin was positive in the mouse lymphoma mutagenesis assay and was clastogenic in the in vivo rat micronucleus assay. No carcinogenicity studies have been conducted with eribulin. A fertility study was not conducted with eribulin, but based on non-clinical findings in repeated-dose studies where testicular toxicity was observed in both rats (hypocellularity of seminiferous epithelium with hypospermia/aspermia) and dogs, male fertility may be compromised by treatment with eribulin. An embryofoetal development study in rat confirmed the developmental toxicity and teratogenic potential of eribulin. Pregnant rats were treated with eribulin mesilate equivalent to 0.009, 0.027, 0.088 and 0.133 mg/kg eribulin at gestation days 8, 10 and 12. Dose related increased number of resorptions and decreased foetal weight were observed at doses ≥ 0.088 mg/kg and increased incidence of malformations (absence of lower jaw, tongue, stomach and spleen) was recorded at 0.133 mg/kg. 6. Pharmaceutical particulars 6.1 List of excipients Ethanol anhydrous Water for injections Hydrochloric acid (for pH-adjustment) Sodium hydroxide (for pH-adjustment) 6.2 Incompatibilities In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products except those mentioned in section 6.6. 6.3 Shelf life Unopened vials 4 years. In-use shelf life From a microbiological point of view unless the method of opening precludes the risk of microbial contamination the product should be used immediately. If not used immediately, in-use storage times and conditions are the responsibility of the user. If not used immediately HALAVEN as the undiluted solution in a syringe should not normally be stored longer than 4 hours at 25°C and ambient lighting, or 24 hours at 2°C - 8°C. Diluted solutions of HALAVEN (0.018 mg/ml to 0.18 mg/ml eribulin in sodium chloride 9 mg/ml (0.9%)) solution for injection should not be stored longer than 24 hours at 2°C - 8°C, unless dilution has taken place in controlled and validated aseptic conditions. 6.4 Special precautions for storage This medicinal product does not require any special storage conditions. For storage conditions after first opening or dilution of the medicinal product, see section 6.3. 6.5 Nature and contents of container 5 ml type I glass vial, with teflon-coated, butyl rubber stopper and flip-off aluminium over seal, containing 2 ml of solution. The pack sizes are cartons of 1 or 6 vials. Not all pack sizes may be marketed. 6.6 Special precautions for disposal and other handling HALAVEN is a cytotoxic anticancer medicinal product and, as with other toxic compounds, caution should be exercised in its handling. The use of gloves, goggles, and protective clothing is recommended. If the skin comes into contact with the solution it should be washed immediately and thoroughly with soap and water. If it contacts mucous membranes, the membranes should be flushed thoroughly with water. HALAVEN should only be prepared and administered by personnel appropriately trained in handling of cytotoxic agents. Pregnant staff should not handle HALAVEN. Using aseptic technique HALAVEN can be diluted up to 100 ml with sodium chloride 9 mg/ml (0.9%) solution for injection. It must not be mixed with other medicinal products and should not be diluted in glucose 5% infusion solution. Any unused medicinal product or waste material should be disposed of in accordance with local requirements. 7. Marketing authorisation holder Eisai Europe Ltd European Knowledge Centre Mosquito Way Hatfield Hertfordshire AL10 9SN United Kingdom 8. Marketing authorisation number(s) EU/1/11/678/001-002 9. Date of first authorisation/renewal of the authorisation Date of first authorisation: 17 March 2011 10. Date of revision of the text 06/2014 Detailed information on this medicinal product is available on the website of the European Medicines Agency http://www.ema.europa.eu CHMP建议批准卫材Halaven乳腺癌新适应症 卫材(Eisai)2014年5月27日宣布,药物Halaven(eribulin)获得了欧洲药品管理局(EMA)人用医药产品委员会(CHMP)的积极意见。CHMP建议批准Halaven用于既往至少接受过一种晚期化疗方案但病情恶化的局部晚期或转移性乳腺癌(MBC)的治疗。既往化疗方案应包括一种蒽环类药物和一种紫衫烷用于辅助疗法或转移性病情,触发患者不适合这些药物。欧盟委员会(EC)预计将在3个月内对Halaven的新适应症做出最终审查决定。经过与FDA的讨论,卫材没有提交Halaven的扩大适应症申请。 CHMP的积极意见,是基于2个全球性III期试验EMBRACE和Study-301的临床证据,这些研究涉及超过1800例患者。 EMBRACE研究显示,与接受医生选择的一种替代方案相比,Halaven能够在很大程度上延长既往过度治疗的转移性乳腺癌患者的平均总生存期(13.2个月 vs 10.5个月,p=0.014)。EMBRACE研究是过去40年中证实能够显著延长总生存期的25项研究之一。 Study-301是一种头对头研究,将Halaven与卡培他滨进行了比较。数据表明,在意向性治疗群体中,与卡培他滨相比,Halaven在改善总生存期(OS)方面显示出有利的趋势,尽管改善无统计学显著差异(15.9个月 vs 14.5个月,p=0.056)。 此前,Halaven已在美国、欧盟、瑞士、日本、新加坡获批,用于先前因癌症转移而接受至少2次化疗以及既往疗法包括蒽环类和紫杉类化疗药物的乳腺癌患者的治疗。 Halaven是一种合成的大田软海绵素(halichondrin B)类似物。Halichondrin B是一种从生长在日本沿海的黑色海绵上发现的物质,能够有效治愈肿瘤。Halaven于2010年在美国上市,迄今全球共有4.9万例患者接受Halaven治疗。 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||