|
近日,欧盟委员会(EC)批准了全球首个批准的前蛋白转化酶枯草溶菌素/kexin 9型(PCSK9)抑制剂RepathaTM (evolocumab)的上市许可,该药批准用于胆固醇控制不良而需要额外强化降低低密度脂蛋白胆固醇(LDL-C)的患者.
Repatha PFS
Amgen Ltd
1. Name of the medicinal product
Repatha® 140 mg solution for injection in pre-filled syringe
2. Qualitative and quantitative composition
Each pre-filled syringe contains 140 mg of evolocumab in 1 mL of solution.
Repatha is a human IgG2 monoclonal antibody produced using recombinant DNA technology in Chinese hamster ovary (CHO) cells.
For the full list of excipients, see section 6.1.
3. Pharmaceutical form
Solution for injection (injection).
The solution is clear to opalescent, colourless to yellowish, and practically free from particles.
4. Clinical particulars
4.1 Therapeutic indications
Hypercholesterolaemia and mixed dyslipidaemia
Repatha is indicated in adults with primary hypercholesterolaemia (heterozygous familial and non-familial) or mixed dyslipidaemia, as an adjunct to diet:
• in combination with a statin or statin with other lipid lowering therapies in patients unable to reach LDL-C goals with the maximum tolerated dose of a statin or,
• alone or in combination with other lipid-lowering therapies in patients who are statin-intolerant, or for whom a statin is contraindicated.
Homozygous familial hypercholesterolaemia
Repatha is indicated in adults and adolescents aged 12 years and over with homozygous familial hypercholesterolaemia in combination with other lipid-lowering therapies.
The effect of Repatha on cardiovascular morbidity and mortality has not yet been determined.
4.2 Posology and method of administration
Prior to initiating Repatha, secondary causes of hyperlipidaemia or mixed dyslipidaemia (e.g., nephrotic syndrome, hypothyroidism) should be excluded.
Posology
Primary hypercholesterolaemia and mixed dyslipidaemia in adults
The recommended dose of Repatha is either 140 mg every two weeks or 420 mg once monthly; both doses are clinically equivalent.
Homozygous familial hypercholesterolaemia in adults and adolescents aged 12 years and over
The initial recommended dose is 420 mg once monthly. After 12 weeks of treatment, dose frequency can be up-titrated to 420 mg once every 2 weeks if a clinically meaningful response is not achieved. Patients on apheresis may initiate treatment with 420 mg every two weeks to correspond with their apheresis schedule.
Patients with renal impairment
No dose adjustment is necessary in patients with mild to moderate renal impairment, see section 4.4 for patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2).
Patients with hepatic impairment
No dose adjustment is necessary in patients with mild hepatic impairment, see section 4.4 for patients with moderate and severe hepatic impairment.
Elderly patients (age ≥ 65 years)
No dose adjustment is necessary in elderly patients.
Paediatric population
The safety and efficacy of Repatha in children aged less than 18 years has not been established in the indication for primary hypercholesterolaemia and mixed dyslipidaemia. No data are available.
The safety and efficacy of Repatha in children aged less than 12 years has not been established in the indication for homozygous familial hypercholesterolaemia. No data are available.
Method of administration
Subcutaneous use.
Repatha is for subcutaneous injection into the abdomen, thigh or upper arm region. Injection sites should be rotated and injections should not be given into areas where the skin is tender, bruised, red, or hard. Repatha must not be administered intravenously or intramuscularly.
The 420mg dose once monthly or every 2 weeks should be delivered using three pre-filled syringes administered consecutively within 30 minutes.
Repatha is intended for patient self-administration after proper training. Administration of Repatha can also be performed by an individual who has been trained to administer the product.
Each pre-filled syringe is for single use only.
For instructions on administration, see section 6.6 and the 'Instructions for Use' provided in the carton.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
4.4 Special warnings and precautions for use
Renal impairment
Patients with severe renal impairment (defined as eGFR < 30 mL/min/1.73 m2) have not been studied (see section 5.3). Repatha should be used with caution in patients with severe renal impairment.
Hepatic impairment
In patients with moderate hepatic impairment, a reduction in total evolocumab exposure was observed that may lead to a reduced effect on LDL-C reduction. Therefore, close monitoring may be warranted in these patients.
Patients with severe hepatic impairment (Child-Pugh C) have not been studied (see section 5.3). Repatha should be used with caution in patients with severe hepatic impairment.
Dry natural rubber
The needle cover of the glass pre-filled syringe is made from dry natural rubber (a derivative of latex), which may cause allergic reactions.
Sodium content
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, i.e. it is essentially 'sodium-free'.
4.5 Interaction with other medicinal products and other forms of interaction
No formal drug-drug interaction studies have been conducted for Repatha.
The pharmacokinetic interaction between statins and evolocumab was evaluated in the Repatha clinical trials. An approximately 20% increase in the clearance of evolocumab was observed in patients co-administered statins. This increased clearance is in part mediated by statins increasing the concentration of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) which did not adversely impact the pharmacodynamic effect of evolocumab on lipids. No statin dose adjustments are necessary when used in combination with Repatha.
No studies on pharmacokinetic and pharmacodynamics interaction between Repatha and lipid-lowering drugs other than statins and ezetimibe have been conducted.
4.6 Fertility, pregnancy and lactation
Pregnancy
There are no or limited amount of data from the use of Repatha in pregnant women.
Animal studies do not indicate direct or indirect effects with respect to reproductive toxicity (see section 5.3).
Repatha should not be used during pregnancy unless the clinical condition of the woman requires treatment with evolocumab.
Breast-feeding
It is unknown whether evolocumab is excreted in human milk.
A risk to breastfed newborns/infants cannot be excluded.
A decision must be made whether to discontinue breast-feeding or discontinue/abstain from Repatha therapy taking into account the benefit of breast-feeding for the child and the benefit of therapy for the woman.
Fertility
No data on the effect of evolocumab on human fertility are available. Animal studies did not show any effects on fertility endpoints at area under the concentration time curve (AUC) exposure levels much higher than in patients receiving evolocumab at 420 mg once monthly (see section 5.3).
4.7 Effects on ability to drive and use machines
Repatha has no known influence on the ability to drive and use machines.
4.8 Undesirable effects
Summary of the safety profile
The most commonly reported adverse drug reactions during primary hypercholesterolaemia and mixed dyslipidaemia pivotal trials, at the recommended doses, were nasopharyngitis (4.8%), upper respiratory tract infection (3.2%), back pain (3.1%), arthralgia (2.2%), influenza (2.3%), and nausea (2.1%). The safety profile in the homozygous familial hypercholesterolaemia population was consistent with that demonstrated in the primary hypercholesterolaemia and mixed dyslipidaemia population.
Tabulated summary of adverse reactions
Adverse reactions reported in pivotal, controlled clinical studies in patients with primary hypercholesterolaemia and mixed dyslipidaemia and homozygous familial hypercholesterolaemia are displayed by system organ class and frequency in table 1 below using the following convention: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000) and very rare (< 1/10,000).
Table 1. Adverse reactions with Repatha
|
MedDRA system organ class (SOC) |
Adverse reactions |
Frequency category |
|
Infections and infestations |
Influenza |
Common |
|
Nasopharyngitis |
Common |
|
Upper respiratory tract infection |
Common |
|
Immune system disorders |
Rash |
Common |
|
Urticaria |
Uncommon |
|
Gastrointestinal disorders |
Nausea |
Common |
|
Musculoskeletal and connective tissue disorders |
Back pain |
Common |
|
Arthralgia |
Common |
|
General disorders and administration site conditions |
Injection site reactions1 |
Common | 1See section Description of selected adverse reactions
Description of selected adverse reactions
Injection site reactions
The most frequent injection site reactions were injection site erythema, injection site pain and injection site bruising.
Paediatric population
There is limited experience with Repatha in paediatric patients. Fourteen patients aged ≥ 12 to < 18 years with homozygous familial hypercholesterolaemia were included in clinical studies. No difference in safety was observed between adolescent and adult patients with homozygous familial hypercholesterolaemia.
The safety and effectiveness of Repatha in paediatric patients with primary hypercholesterolaemia and mixed dyslipidaemia has not been established.
Elderly population
Although no safety issues were observed in patients over 75 years, data are limited in this age subgroup.
Of the 6026 total number of patients in clinical studies of Repatha, 1779 (30%) were ≥ 65 years old, while 223 (4%) were ≥ 75 years old. No overall differences in safety or efficacy were observed between these patients and younger patients
Immunogenicity
In clinical studies, 0.1% of patients (7 out of 4846 patients with primary hyperlipidaemia and mixed dyslipidaemia and 0 out of 80 patients with homozygous familial hypercholesterolaemia) treated with at least one dose of Repatha tested positive for binding antibody development (4 of these patients had transient antibodies). The patients whose sera tested positive for binding antibodies were further evaluated for neutralising antibodies and none of the patients tested positive for neutralising antibodies. The presence of anti-evolocumab binding antibodies did not impact the pharmacokinetic profile, clinical response, or safety of Repatha.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via:
United Kingdom
Yellow Card Scheme
Website: www.mhra.gov.uk/yellowcard
Ireland
HPRA Pharmacovigilance
Earlsfort Terrace
IRL - Dublin 2
Tel: +353 1 6764971
Fax: +353 1 6762517
Website: www.hpra.ie
e-mail: medsafety@hpra.ie
4.9 Overdose
No adverse effects were observed in animal studies at exposures up to 300-fold higher than those in patients treated with Repatha at 420 mg once monthly.
There is no specific treatment for Repatha overdose. In the event of an overdose, the patient should be treated symptomatically, and supportive measures instituted as required.
5. Pharmacological properties
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Other lipid modifying agents. ATC code: C10AX13
Mechanism of action
Evolocumab binds selectively to PCSK9 and prevents circulating PCSK9 from binding to the low density lipoprotein receptor (LDLR) on the liver cell surface, thus preventing PCSK9-mediated LDLR degradation. Increasing liver LDLR levels results in associated reductions in serum LDL-cholesterol (LDL-C).
Pharmacodynamic effects
In clinical trials, Repatha reduced unbound PCSK9, LDL-C, TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG and Lp(a), and increased HDL-C and ApoA1 in patients with primary hypercholesterolaemia and mixed dyslipidaemia.
A single subcutaneous administration of Repatha 140 mg or 420 mg resulted in maximum suppression of circulating unbound PCSK9 by 4 hours followed by a reduction in LDL-C reaching a mean nadir in response by 14 and 21 days, respectively. Changes in unbound PCSK9 and serum lipoproteins were reversible upon discontinuation of Repatha. No increase in unbound PCSK9 or LDL-C above baseline was observed during the washout of evolocumab suggesting that compensatory mechanisms to increase production of PCSK9 and LDL-C do not occur during treatment.
Subcutaneous regimens of 140 mg every 2 weeks and 420 mg once monthly were equivalent in average LDL-C lowering (mean of weeks 10 and 12) resulting in -72 to -57% from baseline compared with placebo. Treatment with Repatha resulted in a similar reduction of LDL-C when used alone or in combination with other lipid-lowering therapy. The effect of LDL-C lowering is sustained; the longest duration measured was 112 weeks.
Clinical efficacy in primary hypercholesterolaemia and mixed dyslipidaemia
LDL-C reduction of approximately 55% to 75% was achieved with Repatha as early as week 1 and maintained during long-term therapy. Maximal response was generally achieved within 1 to 2 weeks after dosing with 140 mg every 2 weeks and 420 mg once monthly.
In 80-85% of all patients treated with either dose, Repatha demonstrated a ≥ 50% reduction in LDL-C at the mean of weeks 10 and 12. Up to 99% of patients treated with either dose of Repatha achieved an LDL-C of < 2.6 mmol/L and up to 95% achieved an LDL-C < 1.8 mmol/L at the mean of weeks 10 and 12.
Repatha at either dose was effective in all subgroups relative to placebo and ezetimibe, with no notable differences observed between subgroups, such as age, race, gender, region, body-mass index, National Cholesterol Education Program risk, current smoking status, baseline coronary heart disease (CHD) risk factors, family history of premature CHD, glucose tolerance status, (i.e., diabetes mellitus type 2, metabolic syndrome, or neither), hypertension, statin dose and intensity, unbound baseline PCSK9, baseline LDL-C and baseline TG.
Repatha reduced LDL-C, non-HDL-C, Apo B, TC, Lp(a), VLDL-C, TG, TC/HDL-C, and ApoB/ApoA1and increased HDL-C in patients with mixed dyslipidaemia.
Repatha was superior to ezetimibe in reducing LDL-C, TC, ApoB, non-HDL-C, Lp(a), TC/HDL-C, and ApoB/ApoA1.
Combination with a statin and statin with other lipid-lowering therapies
LAPLACE-2 was an international, multicentre, double-blind, randomised, 12-week study in 1896 patients with primary hypercholesterolaemia or mixed dyslipidaemia who were randomised to receive Repatha in combination with statins (rosuvastatin, simvastatin or atorvastatin). Repatha was compared to placebo for the rosuvastatin and simvastatin groups and compared with placebo and ezetimibe for the atorvastatin group.
Repatha significantly reduced LDL-C from baseline to mean of weeks 10 and 12 compared with placebo for the rosuvastatin and simvastatin groups and compared with placebo and ezetimibe for the atorvastatin group (p < 0.001). Repatha significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG and Lp(a) and increased HDL-C from baseline to mean of weeks 10 and 12 as compared to placebo for the rosuvastatin and simvastatin groups (p < 0.05) and significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 and Lp(a), compared with placebo and ezetimibe for the atorvastatin group (p < 0.001) (see tables 2 and 3).
RUTHERFORD-2 was an international, multicentre, double-blind, randomised, placebo-controlled, 12-week study in 329 patients with heterozygous familial hypercholesterolaemia on lipid-lowering therapies. Repatha significantly reduced LDL-C from baseline to mean of weeks 10 and 12 compared with placebo (p < 0.001). Repatha significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 VLDL-C, TG and Lp(a) and increased HDL-C and ApoA1 from baseline to mean of weeks 10 and 12 compared to placebo (p < 0.05) (see table 2).
Table 2: Treatment effects of Repatha compared with placebo in patients with primary hypercholesterolaemia and mixed dyslipidaemia - mean percent change from baseline to average of weeks 10 and 12 (%, CI 95%)
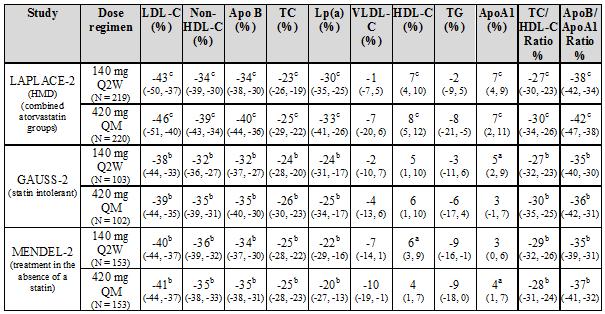
Key: Q2W = once every 2 weeks, QM = once monthly, HMD = Primary hypercholesterolaemia and mixed dyslipidaemia; HeFH = Heterozygous familial hypercholesterolaemia; a p value < 0.05 when compared with placebo. b p value < 0.001 when compared with placebo.
Statin intolerant patients
GAUSS-2 was an international, multicentre, double-blind, randomised, ezetimibe-controlled, 12-week study in 307 patients who were statin-intolerant or unable to tolerate an effective dose of a statin. Repatha significantly reduced LDL-C compared with ezetimibe (p < 0.001). Repatha significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 and Lp(a), from baseline to mean of weeks 10 and 12 compared to ezetimibe (p < 0.001) (see table 3).
Treatment in the absence of a statin
MENDEL-2 was an international, multicentre, double-blind, randomised, placebo and ezetimibe-controlled, 12-week study of Repatha in 614 patients with primary hypercholesterolaemia and mixed dyslipidaemia. Repatha significantly reduced LDL-C from baseline to mean of weeks 10 and 12 compared with both placebo and ezetimibe (p < 0.001). Repatha significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1 and Lp(a), from baseline to mean of weeks 10 and 12 compared with both placebo and ezetimibe (p < 0.001) (see table 3).
Table 3: Treatment effects of Repatha compared with ezetimibe in patients with primary hypercholesterolaemia and mixed dyslipidaemia - mean percent change from baseline to average of weeks 10 and 12 (%, 95% CI)

Key: Q2W = once every 2 weeks, QM = once monthly, HMD = Primary hypercholesterolaemia and mixed dyslipidaemia,a p value < 0.05 when compared with ezetimibe, b p value < 0.001 when compared with ezetimibe, c nominal p value < 0.001 when compared with ezetimibe.
Long-term efficacy in primary hypercholesterolaemia and mixed dyslipidaemia
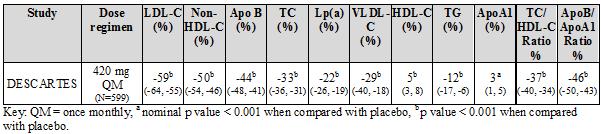
DESCARTES was an international, multicentre, double-blind, randomised, placebo-controlled, 52-week study in 901 patients with hyperlipidaemia who received diet alone, atorvastatin, or a combination of atorvastatin and ezetimibe. Repatha 420 mg once monthly significantly reduced LDL-C from baseline at 52 weeks compared with placebo (p < 0.001). Treatment effects were sustained over 1 year as demonstrated by reduction in LDL-C from week 12 to week 52. Reduction in LDL-C from baseline at week 52 compared with placebo was consistent across background lipid-lowering therapies optimised for LDL-C and cardiovascular risk.
Repatha significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG and Lp(a), and increased HDL-C and ApoA1 at week 52 compared with placebo (p < 0.001) (table 4).
Table 4: Treatment effects of Repatha compared with placebo in patients with primary hypercholesterolaemia and mixed dyslipidaemia - mean percent change from baseline to week 52 (%, 95% CI)
OSLER and OSLER-2 are two ongoing, randomised, controlled, open-label extension studies to assess the long-term safety and efficacy of Repatha in patients who completed treatment in a 'parent' study. In each extension study, patients were randomised 2:1 to receive either Repatha plus standard of care (evolocumab group) or standard of care alone (control group) for the first year of the study. At the end of the first year (week 52 in OSLER and week 48 in OSLER-2), patients were eligible to enter the all Repatha period in which all patients could receive open-label Repatha for either another 4 years (OSLER) or 1 year (OSLER-2).
A total of 1324 patients enrolled in OSLER. Repatha 420 mg once monthly significantly reduced LDL-C from baseline at week 12 and week 52 compared with control (nominal p < 0.001). Treatment effects were maintained over 124 weeks as demonstrated by reduction in LDL-C from week 12 in the parent study to week 112 in the open-label extension. A total of 2928 patients enrolled in OSLER-2. Repatha significantly reduced LDL-C from baseline at week 12 compared with control (nominal p < 0.001). Treatment effects were maintained as demonstrated by reduction in LDL-C from week 12 to week 24 in the open-label extension. Repatha significantly reduced TC, ApoB, non-HDL-C, TC/HDL-C, ApoB/ApoA1, VLDL-C, TG and Lp(a) , and increased HDL-C and ApoA1 from baseline to week 52 in OSLER and to week 24 in OSLER-2 compared with control (nominal p < 0.001). LDL-C and other lipid parameters returned to baseline within 12 weeks after discontinuation of Repatha at the beginning of OSLER or OSLER-2 without evidence of rebound.
TAUSSIG is an ongoing multicentre, open-label, 5-year extension study to assess the long-term safety and efficacy of Repatha, as an adjunct to other lipid lowering therapies, in patients with severe familial hypercholesterolaemia, including homozygous familial hypercholesterolaemia. A total of 102 severe familial hypercholesterolaemia patients and 96 homozygous familial hypercholesterolaemia patients enrolled in TAUSSIG. All patients in the study were initially treated with Repatha 420 mg once monthly, except for those receiving apheresis at enrolment who began with Repatha 420 mg once every 2 weeks. Dose frequency in non-apheresis patients could be titrated up to 420 mg once every 2 weeks based on LDL-C response and PCSK9 levels. Long-term use of Repatha demonstrated a sustained treatment effect as evidenced by reduction of LDL-C in patients with severe familial hypercholesterolaemia (table 5).
Changes in other lipid parameters (TC, ApoB, non-HDL-C, TC/HDL-C, and ApoB/ApoA1) also demonstrated a sustained effect of long-term Repatha administration in patients with severe familial hypercholesterolaemia.
Table 5: Effect of Repatha on LDL-C in patients with severe familial hypercholesterolaemia – median percent change from baseline to OLE week 36
|
Patient population
(N) |
OLE week 12
(n = 16) |
OLE week 24
(n = 8) |
OLE week 36
(n = 5) |
|
Severe FH (N = 102) |
-47 |
-45 |
-48 | Key: OLE = open-label extension, N (n) = Number of evaluable patients (N) and patients with observed LDL values at specific scheduled visit (n) in the severe familial hypercholesterolaemia interim analysis set
The clinical relevance, including the long-term safety, of sustained very low levels of LDL C (i.e., < 0.65 mmol/L [< 25 mg/dL]) have not yet been established. Available data demonstrate that there are no clinically meaningful differences between the safety profiles of patients with LDL-C levels < 0.65 mmol/L and those with higher LDL-C, see section 4.8.
Clinical efficacy in homozygous familial hypercholesterolaemia
TESLA was an international, multicentre, double-blind, randomised, placebo-controlled 12-week study in 49 homozygous familial hypercholesterolemia patients aged 12 to 65 years. Repatha 420 mg once monthly, as an adjunct to other lipid-lowering therapies (e.g., statins, bile-acid sequestrants), significantly reduced LDL-C and ApoB at week 12 compared with placebo (p < 0.001) (table 6). Changes in other lipid parameters (TC, non-HDL-C, TC/HDL-C, and ApoB/ApoA1) also demonstrated a treatment effect of Repatha administration in patients with homozygous familial hypercholesterolemia.
Table 6: Treatment effects of Repatha compared with placebo in patients with homozygous familial hypercholesterolaemia - mean percent change from baseline to week 12 (%, CI 95%)

Key: HoFH = homozygous familial hypercholesterolaemia; QM = once monthly; a nominal p value <0.001 when compared with placebo; b p value < 0.001 when compared with placebo.
Long-term efficacy in homozygous familial hypercholesterolaemia
In TAUSSIG, long-term use of Repatha demonstrated a sustained treatment effect as evidenced by reduction of LDL-C of approximately 20% to 30% in patients with homozygous familial hypercholesterolaemia not on apheresis and approximately 15% to 25% in patients with homozygous familial hypercholesterolaemia on apheresis (table 7). Changes in other lipid parameters (TC, ApoB, non-HDL-C, TC/HDL-C, and ApoB/ApoA1) also demonstrated a sustained effect of long-term Repatha administration in patients with homozygous familial hypercholesterolaemia. Reductions in LDL-C and changes in other lipid parameters in 13 adolescent patients (aged ≥ 12 to < 18 years) with homozygous familial hypercholesterolaemia are comparable to those in the overall population of patients with homozygous familial hypercholesterolaemia.
Table 7: Effect of Repatha on LDL-C in patients with homozygous familial hypercholesterolaemia - Mean percent change from baseline to OLE week 36
|
Patient population
(N) |
OLE week 12 |
OLE week 24 |
OLE week 36 |
|
HoFH
(N = 96) |
-20
(n = 70) |
-23
(n = 46) |
-24
(n = 30) |
|
Non-apheresis
(N = 65) |
-22
(n = 46) |
-24
(n = 33) |
-24
(n = 27) |
|
Apheresis
(N = 31) |
-17
(n = 24) |
-20
(n = 13) |
-21
(n = 3) | Key: OLE = open label extension. N (n) = Number of evaluable patients (N) and patients with observed LDL values at specific schedule visit (n) in the HoFH interim analysis set
The effect of Repatha on cardiovascular morbidity and mortality has not yet been demonstrated.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Repatha in all subsets of the paediatric population in the treatment of mixed dyslipidaemia.
The European Medicines Agency has deferred the obligation to submit the results of studies with Repatha in one or more subsets of the paediatric population in the treatment of elevated cholesterol.
There are limited data available on the use of Repatha in the paediatric population. Fourteen adolescent patients aged ≥ 12 to < 18 years with homozygous familial hypercholesterolaemia have been included in clinical trials. No overall differences in safety or efficacy were observed between adolescent and adult patients with homozygous familial hypercholesterolaemia.
See section 4.2 for information on paediatric use.
5.2 Pharmacokinetic properties
Absorption and distribution
Following a single subcutaneous dose of 140 mg or 420 mg Repatha administered to healthy adults, median peak serum concentrations were attained in 3 to 4 days. Administration of single subcutaneous dose of 140 mg resulted in a Cmax mean (SD) of 13.0 (10.4) μg/mL and AUClast mean (SD) of 96.5 (78.7) day•μg/mL. Administration of single subcutaneous dose 420 mg resulted in a Cmax mean (SD) of 46.0 (17.2) μg/mL and AUClast mean (SD) of 842 (333) day•μg/mL. Three subcutaneous 140 mg doses were bioequivalent to a single subcutaneous 420 mg dose. The absolute bioavailability after SC dosing was determined to be 72% from pharmacokinetic models.
Following a single 420 mg Repatha intravenous dose, the mean (SD) steady-state volume of distribution was estimated to be 3.3 (0.5) L, suggesting evolocumab has limited tissue distribution.
Biotransformation
Repatha is composed solely of amino acids and carbohydrates as native immunoglobulin and is unlikely to be eliminated via hepatic metabolic mechanisms. Its metabolism and elimination are expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides and individual amino acids.
Elimination
Evolocumab was estimated to have an effective half-life of 11 to 17 days.
In patients with heterozygous familial hypercholesterolemia (HeFH) on high dose statin, the systemic exposure of evolocumab was slightly lower than in subjects on low-to-moderate dose statin (the ratio of AUClast 0.74 [90%CI 0.29 ; 1.9]). An approximately 20% increase in the clearance is in part mediated by statins increasing the concentration of PCSK9 which did not adversely impact the pharmacodynamic effect of evolocumab on lipids. Population pharmacokinetic analysis indicated no appreciable differences in evolocumab serum concentrations in hypercholesterolaemic patients (non-familial hypercholesterolaemia or familial hypercholesterolaemia) taking concomitant statins.
Linearity/non-linearity
Following a single 420 mg intravenous dose, the mean (SD) systemic clearance was estimated to be 12 (2) mL/hr. In clinical studies with repeated subcutaneous dosing over 12 weeks, dose proportional increases in exposure were observed with dose regimens of 140 mg and greater. An approximate two to three-fold accumulation was observed in trough serum concentrations (Cmin (SD) 7.21 (6.6)) following 140 mg doses every 2 weeks or following 420 mg doses administered monthly (Cmin (SD) 11.2 (10.8)), and serum trough concentrations approached steady-state by 12 weeks of dosing.
No time dependent changes were observed in serum concentrations over a period of 124 weeks.
Renal impairment
No dose adjustment is necessary in patients with mild to moderate renal impairment. Population pharmacokinetic analysis of integrated data from the Repatha clinical trials did not reveal a difference in pharmacokinetics of evolocumab in patients with mild or moderate renal impairment relative to non-renally impaired patients. Repatha has not been studied in patients with severe renal impairment (see section 4.4).
Hepatic impairment
No dose adjustment is necessary in patients with mild hepatic impairment (Child-Pugh class A). Single 140 mg subcutaneous doses of Repatha were studied in 8 patients with mild hepatic impairment, 8 patients with moderate hepatic impairment and 8 healthy subjects. The exposure to evolocumab was found to be approximately 40-50% lower compared to healthy subjects. However, baseline PCSK9 levels and the degree and time course of PCSK9 neutralisation were found to be similar between patients with mild or moderate hepatic impairment and healthy volunteers. This resulted in similar time course and extent of absolute LDL-C lowering. Repatha has not been studied in patients with severe hepatic impairment (Child-Pugh class C) (see section 4.4).
Body Weight
Body weight was a significant covariate in population PK analysis impacting evolocumab trough concentrations, however there was no impact on LDL-C reduction. Following repeat subcutaneous administration of 140 mg every 2 weeks, the 12 week trough concentrations were 147% higher and 70% lower in patients of 69 kg and 93 kg respectively, than that of the typical 81 kg subject. Less impact from body weight was seen with repeated subcutaneous evolocumab 420 mg monthly doses.
Other special populations
Population pharmacokinetic analyses suggest that no dose adjustments are necessary for age, race or gender. The pharmacokinetics of evolocumab were influenced by body weight without having any notable effect on LDL-C lowering. Therefore, no dose adjustments are necessary based on body weight.
5.3 Preclinical safety data
Evolocumab was not carcinogenic in hamsters at exposures much higher than patients receiving evolocumab at 420 mg once monthly. The mutagenic potential of evolocumab has not been evaluated.
In hamsters and cynomolgus monkeys at exposures much higher than patients receiving 420 mg evolocumab once monthly, no effect on male or female fertility was observed.
In cynomolgus monkeys at exposures much higher than patients receiving 420 mg evolocumab once monthly, no effects on embryo-foetal or postnatal development (up to 6 months of age) were observed.
Apart from a reduced T-cell Dependent Antibody Response in cynomolgus monkeys immunized with KLH after 3 months of treatment with evolocumab, no adverse effects were observed in hamsters (up to 3 months) and cynomolgus monkeys (up to 6 months) at exposures much higher than patients receiving evolocumab at 420 mg once monthly. The intended pharmacological effect of decreased serum LDL-C and total cholesterol were observed in these studies and was reversible upon cessation of treatment.
In combination with rosuvastatin for 3 months, no adverse effects were observed in cynomolgous monkeys at exposures much higher than patients receiving 420 mg evolocumab once monthly. Reductions in serum LDL-C and total cholesterol were more pronounced than observed previously with evolocumab alone, and were reversible upon cessation of treatment.
6. Pharmaceutical particulars
6.1 List of excipients
Proline
Glacial acetic acid
Polysorbate 80
Sodium hydroxide (for pH adjustment)
Water for injections
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.3 Shelf life
2 years.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C). Do not freeze.
Keep the pre-filled syringe in the original carton in order to protect from light.
If removed from the refrigerator, Repatha may be stored at room temperature (up to 25°C) in the original carton and must be used within 1 week.
6.5 Nature and contents of container
One ml solution in a single use pre-filled syringe made from type I glass with stainless steel 27 gauge needle.
The needle cover of the pre-filled syringe is made from dry natural rubber (a derivative of latex, see section 4.4).
Pack size of one pre-filled syringe.
6.6 Special precautions for disposal and other handling
Before administration, the solution should be inspected. Do not inject the solution if it contains particles, or is cloudy or discoloured. To avoid discomfort at the site of injection, allow the pre-filled syringe to reach room temperature (up to 25°C) before injecting. Inject the entire contents of the pre-filled syringe.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
7. Marketing authorisation holder
Amgen Europe B.V.
Minervum 7061
4817 ZK Breda
The Netherlands
8. Marketing authorisation number(s)
EU/1/15/1016/001
9. Date of first authorisation/renewal of the authorisation
Date of first authorisation: 17 July 2015
10. Date of revision of the text
July 2015
Detailed information on this medicinal product is available on the website of the European Medicines Agency http://www.ema.europa.eu.

全球首个PCSK9抑制剂--安进新型降胆固醇REPATHA用于高胆固醇治疗
2015年7月24日,欧盟委员会(EC)批准全球首个前蛋白转化酶枯草溶菌素/kexin 9型(PCSK9)抑制剂RepathaTM(evolocumab)的上市许可,批准该药用于胆固醇控制不良而需要额外强化降低低密度脂蛋白胆固醇(LDL-C)的患者。Repatha是一种可抑制PCSK9的全人源化单克隆抗体,PCSK9是一种能够降低肝脏从血液中清除LDL-C或“坏”胆固醇的能力的蛋白。LDL-C升高是血液中胆固醇和/或脂肪发生异常2,3,被视为心血管疾病 (CVD) 的重要风险因素。
EC已批准Repatha用于:
作为饮食的一种辅助疗法,用于治疗原发性高胆固醇血症(杂合子家族性和非家族性[HeFH])或混合性血脂异常成年患者:联合他汀类药物、或者联合他汀类及其他降脂疗法,用于接受最大耐受剂量的他汀类药物治疗仍无法达到LDL-C目标的患者,或者单独或联合其他降脂疗法,用于不能耐受他汀或者他汀类药物禁忌的患者。联合其他降脂疗法,用于成人或12岁及以上年龄的纯合子家族性(HoFH)患者。
Repatha对心血管发病率和死亡率的影响尚未确定。
欧洲超过60%的高危患者在使用他汀或其他当前获批的降脂药物时仍无法充分降低其LDL-C水平。在极高危患者中,这一比例增至80%以上。欧盟每年CVD方面的医疗保健成本大约为1060亿欧元。
“我们的降胆固醇药物Repatha是全球监管当局批准的首个PCSK9抑制剂,我们为此感到自豪”。安进研发执行副总裁、医学博士Sean E. Harper表示,“LDL胆固醇升高是一项重大的全球健康负担,很多患者在使用最大耐受剂量他汀时仍不能适当控制其LDL胆固醇水平,或因不耐受或禁忌而不能使用他汀类药物。能让欧洲患者获得这种新型降胆固醇药物,这令我们很激动。”
家族性高胆固醇血症 (FH) 患者是一类高危患者群体。FH是一种因基因突变造成的、可在幼年时导致高水平LDL-C的遗传性疾病8。估计在大部分国家中,只有低于1%的FH患者(杂合子和纯合子)被确诊。
“很多接受降胆固醇治疗的患者,包括家族性高胆固醇血症患者,仍在努力控制他们的LDL胆固醇水平”,阿姆斯特丹大学学术医疗中心(AMC) 血管医学科主任J.P.Kastelein医学教授表示,“作为欧盟此类新型药物的首位成员,对于胆固醇控制不良而需要额外降低LDL胆固醇的患者,evolocumab将为医师提供一个重要的、创新的治疗选择。”
EC的批准授予了欧盟28个成员国使用统一标签的集中上市许可。作为欧洲经济区 (EEA) 成员,挪威、冰岛和列支敦斯登将基于EC的决定作出相应决策。
数据显示,Repatha已在大约6000例原发性高脂血症和混合性血脂异常患者(包括10项3期研究中超过4500例高胆固醇患者在内)中证实了显著且一致的降低LDL-C水平的作用以及其他脂质参数支持性的获益变化。在这些研究中,Repatha较安慰剂显著降低LDL-C水平大约达55-75%;Repatha较依折麦布显著降低LDL-C水平大约达35-45%11,12,14。在纯合子FH患者中,Repatha较安慰剂显著降低LDL-C的水平大约达15-30%15。通过长期治疗可维持LDL-C的下降。
Repatha的不良事件属性总体上与对照组相似11-17。Repatha组发生率≥2%、且频率高于对照组的最常见的不良事件包括鼻咽炎、上呼吸道感染、背痛、关节痛、流感和恶心。完整安全性信息请参见产品特性总结 (SmPC)。
Repatha可在腹部、大腿和上臂区域皮下注射使用。注射部位应轮流交替,不应在娇嫩、擦伤、红肿或硬结皮肤区域注射药物。禁止静脉或肌肉注射给予Repatha。在启动Repatha 治疗前,应排除导致血液中胆固醇过高和脂肪水平异常的继发性因素(非遗传性)。本品仅可通过处方获得。
原发性高胆固醇血症患者成人推荐剂量为140mg每两周一次或420mg(3支预充注射器)每月一次;两种剂量具有临床等效性。对于成人纯合子FH患者或大于12岁的儿童纯合子FH患者,起始推荐剂量为420mg每月一次。如经过12周治疗后仍未能实现缓解,剂量可增至420mg每两周一次。更多信息请参见包装说明书。
关于RepathaTM (evolocumab)
RepathaTM (evolocumab) 是一种可抑制前蛋白转化酶枯草溶菌素/kexin9型 (PCSK9) 的全人源化单克隆抗体1。PCSK9是一种能够降解LDL受体,从而降低肝脏从血液中清除LDL-C或“坏”胆固醇的能力的蛋白18。Repatha由安进的科学家研发获得,用于结合PCSK9,从而抑制PCSK9与肝脏表面LDL受体的结合。在PCSK9缺失的条件下,肝脏表面的LDL受体水平更高,从而能够更有效地清除血液中的LDL-C1。
重要欧盟产品信息
高胆固醇血症和混合性血脂异常
Repatha适用于作为饮食的辅助疗法,治疗原发性高胆固醇血症(杂合子家族性和非家族性[HeFH])或混合性血脂异常成年患者:
联合他汀类药物、或者联合他汀类及其他降脂疗法,用于接受最大耐受剂量的他汀类药物治疗仍无法达到LDL-C目标的患者,或者单独或联合其他降脂疗法,用于不能耐受或者他汀类药物禁忌的患者。
纯合子家族性高胆固醇血症
Repatha适用于联合其他降脂疗法,用于成人或12岁及以上年龄的纯合子家族性(HoFH)患者。
Repatha对心血管发病率和死亡率的影响尚未确定。
重要安全性信息
对本品可进行额外监测。这样做可迅速鉴定出新的安全性信息。要求医疗专业人士报告所有可疑的不良反应。
禁忌症:
对活性物质或任何辅料过敏。
特殊警告和注意事项:
肾功能损害:
尚未对重度肾功能受损(定义为eGFR<30mL/min/1.73m2)患者进行研究。在重度肾功能受损患者中应谨慎使用Repatha。 肝功能损害:在中度肝功能受损患者中观察到evolocumab总暴露量下降,可能导致降LDL-C作用减弱。因此需对此类患者进行密切监测。尚未对重度肝功能受损(Child-Pugh C)患者进行研究。在重度肝功能受损患者中应谨慎使用Repatha。干天然橡胶:玻璃预充注射器和预充笔的针帽材质为干天然橡胶(或乳胶衍生物),可能导致过敏反应。钠含量:Repatha每剂含钠量低于1mmol(23mg),即基本不含钠。
相互作用:
尚未对Repatha进行药物相互作用研究。未对Repatha与他汀类药物和依泽麦布以外降脂药物之间的药代动力学和药效学相互作用进行研究。
生育能力、妊娠及哺乳:
在妊娠妇女中使用Repatha的数据有限。不应在妊娠妇女中使用Repatha,除非此类患者临床上需要使用evolocumab治疗。尚不清楚evolocumab能否经人乳分泌。尚不能排除对母乳喂养新生儿/婴儿的风险。尚无evolocumab对人生育能力影响的数据。
不良影响:
在关键性对照临床研究中报告了下列常见不良反应(≥ 1/100-< 1/10):流感、鼻咽炎、上呼吸道感染、皮疹、恶心、背痛、关节痛、注射部位反应。不良反应的完整描述请参见SmPC。
药品注意事项:
冰箱内保存(2–8 摄氏度),不得冷冻。应将预充注射器和预充笔置于原包装纸箱内以避光。如从冰箱内取出,Repatha可在室温下(最高25 摄氏度)置于原包装纸箱内保存,但必须在1周内使用。
|
