作成又は改訂年月
2011年1月作成(第1版)
日本標準商品分類番号
873949
日本標準商品分類番号等
国際誕生年月
2008年4月
薬効分類名
非プリン型選択的キサンチンオキシダーゼ阻害剤
高尿酸血症治療剤
承認等
販売名
フェブリク錠10mg
販売名コード
3949003F1023
承認・許可番号
承認番号
22300AMX00419000
商標名
Feburic Tablet 10mg
薬価基準収載年月
2011年3月
販売開始年月
2011年5月
貯法・使用期限等
貯法
室温保存
使用期限
外箱に表示
規制区分
処方せん医薬品注)
注)注意-医師等の処方せんにより使用すること
組成
有効成分・名称
フェブキソスタット
有効成分・含量(1錠中)
10mg
添加物
乳糖、部分アルファー化デンプン、ヒドロキシプロピルセルロース、クロスカルメロースナトリウム、ステアリン酸マグネシウム、ヒプロメロース、マクロゴール
性状
剤形
錠剤
色調・性状
白色~微黄色、円形のフィルムコーティング錠
外形・表面

外形・裏面

外形・側面

大きさ・直径
約7mm
大きさ・厚さ
約3mm
質量
約132mg
識別コード
TJN FET:10
販売名
フェブリク錠20mg
販売名コード
3949003F2020
承認・許可番号
承認番号
22300AMX00421000
商標名
Feburic Tablet 20mg
薬価基準収載年月
2011年3月
販売開始年月
2011年5月
貯法・使用期限等
貯法
室温保存
使用期限
外箱に表示
規制区分
処方せん医薬品注)
注)注意-医師等の処方せんにより使用すること
組成
有効成分・名称
フェブキソスタット
有効成分・含量(1錠中)
20mg
添加物
乳糖、部分アルファー化デンプン、ヒドロキシプロピルセルロース、クロスカルメロースナトリウム、ステアリン酸マグネシウム、ヒプロメロース、マクロゴール
性状
剤形
錠剤
色調・性状
白色~微黄色、円形の割線入りフィルムコーティング錠
外形・表面

外形・裏面

外形・側面

大きさ・直径
約7mm
大きさ・厚さ
約3mm
質量
約132mg
識別コード
TJN FET:20
販売名
フェブリク錠40mg
販売名コード
3949003F302
承認・許可番号
承認番号
22300AMX00420000
商標名
Feburic Tablet 40mg
薬価基準収載年月
2011年3月
販売開始年月
2011年5月
貯法・使用期限等
貯法
室温保存
使用期限
外箱に表示
規制区分
処方せん医薬品注)
注)注意-医師等の処方せんにより使用すること
組成
有効成分・名称
フェブキソスタット
有効成分・含量(1錠中)
40mg
添加物
乳糖、部分アルファー化デンプン、ヒドロキシプロピルセルロース、クロスカルメロースナトリウム、ステアリン酸マグネシウム、ヒプロメロース、マクロゴール
性状
剤形
錠剤
色調・性状
白色~微黄色、円形の割線入りフィルムコーティング錠
外形・表面

外形・裏面

外形・側面

大きさ・直径
約9mm
大きさ・厚さ
約4mm
質量
約261mg
識別コード
TJN FET:40
一般的名称
フェブキソスタット製剤
禁忌
(次の患者には投与しないこと)
1.
本剤の成分に対し過敏症の既往歴のある患者
2.
メルカプトプリン水和物又はアザチオプリンを投与中の患者[「相互作用」の項参照]
|
効能又は効果
痛風、高尿酸血症
効能又は効果に関連する使用上の注意
1.
本剤の適用にあたっては、最新の治療指針等を参考に、薬物治療が必要とされる患者を対象とすること。
2.
女性患者における安全性及び有効性は確立していない。[使用経験が少ない。]
用法及び用量
通常、成人にはフェブキソスタットとして1日10mgより開始し、1日1回経口投与する。その後は血中尿酸値を確認しながら必要に応じて徐々に増量する。維持量は通常1日1回40mgで、患者の状態に応じて適宜増減するが、最大投与量は1日1回60mgとする。
用法及び用量に関連する使用上の注意
尿酸降下薬による治療初期には、血中尿酸値の急激な低下により痛風関節炎(痛風発作)が誘発されることがあるので、本剤の投与は10mg1日1回から開始し、投与開始から2週間以降に20mg1日1回、投与開始から6週間以降に40mg1日1回投与とするなど、徐々に増量すること(「臨床成績」の項参照)。なお、増量後は経過を十分に観察すること。
使用上の注意
慎重投与
(次の患者には慎重に投与すること)
1.
重度の腎機能障害のある患者[使用経験が少なく安全性が確立していない。]
2.
肝機能障害のある患者[使用経験が少なく安全性が確立していない。]
重要な基本的注意
1.
本剤は尿酸降下薬であり、痛風関節炎(痛風発作)発現時に血中尿酸値を低下させると痛風関節炎(痛風発作)を増悪させるおそれがあるため、本剤投与前に痛風関節炎(痛風発作)が認められた場合は、症状がおさまるまで、本剤の投与を開始しないこと。また、本剤投与中に痛風関節炎(痛風発作)が発現した場合には、本剤の用量を変更することなく投与を継続し、症状によりコルヒチン、非ステロイド性抗炎症剤、副腎皮質ステロイド等を併用すること
2.
本剤投与中は甲状腺関連の所見の有無を確認し、異常が認められた場合には甲状腺機能関連の検査を実施すること。
相互作用
併用禁忌
(併用しないこと)
薬剤名等
メルカプトプリン水和物(ロイケリン)
アザチオプリン(イムラン、アザニン)
臨床症状・措置方法
骨髄抑制等の副作用を増強する可能性がある。
機序・危険因子
アザチオプリンの代謝物メルカプトプリンの代謝酵素であるキサンチンオキシダーゼの阻害により、メルカプトプリンの血中濃度が上昇することがアロプリノール(類薬)で知られている。本剤もキサンチンオキシダーゼ阻害作用をもつことから、同様の可能性がある。
|
併用注意
(併用に注意すること)
薬剤名等
ビダラビン
臨床症状・措置方法
幻覚、振戦、神経障害等のビダラビンの副作用を増強する可能性がある。
機序・危険因子
ビダラビンの代謝酵素であるキサンチンオキシダーゼの阻害により、ビダラビンの代謝を抑制し、作用を増強させることがアロプリノール(類薬)で知られている。本剤もキサンチンオキシダーゼ阻害作用をもつことから、同様の可能性がある。
薬剤名等
ジダノシン
臨床症状・措置方法
ジダノシンの血中濃度が上昇する可能性がある。
本剤と併用する場合は、ジダノシンの投与量に注意すること。
機序・危険因子
ジダノシンの代謝酵素であるキサンチンオキシダーゼの阻害により、健康成人及びHIV患者においてジダノシンのCmax及びAUCが上昇することがアロプリノール(類薬)で知られている。本剤もキサンチンオキシダーゼ阻害作用をもつことから、同様の可能性がある。
副作用
副作用等発現状況の概要
承認時までの安全性評価対象1,027例中228例(22.2%)に副作用(臨床検査値の異常を含む)が認められた。内訳は、自他覚的副作用が80例(7.8%)、臨床検査値異常が81例(7.9%)、痛風関節炎は105例(10.2%)であった。主な自他覚的副作用は関節痛12例(1.2%)、四肢不快感9例(0.9%)、四肢痛9例(0.9%)、下痢8例(0.8%)、倦怠感5例(0.5%)等であった。副作用とされた臨床検査値の異常は、肝機能検査値異常36例(3.5%)、TSH増加9例(0.9%)、尿中β2ミクログロブリン増加8例(0.8%)、CK(CPK)増加5例(0.5%)等であった。
重大な副作用
1. 肝機能障害
(頻度不明)
AST(GOT)、ALT(GPT)等の上昇を伴う肝機能障害があらわれることがあるので、本剤投与中は定期的に検査を行うなど、患者の状態を十分に観察し、異常が認められた場合には適切な処置を行うこと。
2. 過敏症
(頻度不明)
全身性皮疹、発疹などの過敏症があらわれることがあるので、観察を十分に行い、異常が認められた場合には適切な処置を行うこと。
その他の副作用
1. 内分泌系
1%未満
TSH増加
2. 神経系
頻度不明
頭痛注1)
3. 神経系
1%未満
手足のしびれ感、浮動性めまい、傾眠
4. 心臓
1%未満
心電図異常
5. 胃腸
1%未満
下痢、腹部不快感、悪心、腹痛
6. 肝・胆道系
1~5%未満
肝機能検査値異常〔ALT(GPT)増加、AST(GOT)増加、γ-GTP増加等〕
7. 皮膚
1%未満
発疹
8. 筋骨格系
1~5%未満
関節痛
9. 筋骨格系
1%未満
四肢痛、四肢不快感、CK(CPK)増加
10. 腎及び尿路
1%未満
β-NアセチルDグルコサミニダーゼ増加、尿中β2ミクログロブリン増加
11. その他
1%未満
倦怠感、口渇、血中トリグリセリド増加、CRP増加
その他の副作用の注意
以上の副作用が認められた場合には、症状に応じて減量、投与中止などの適切な処置を行うこと。
注1)国内の臨床試験では認められず、外国において認められている。
高齢者への投与
一般に高齢者では生理機能が低下していることが多いため、患者の状態を観察し、十分に注意しながら本剤を投与すること。
妊婦、産婦、授乳婦等への投与
1.
妊婦又は妊娠している可能性のある婦人には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。[妊娠中の投与に関する安全性は確立していない。]
2.
授乳中の婦人には、本剤投与中は授乳を避けさせること。[動物実験(ラット)で本剤が乳汁中に移行することが報告されている。また、動物実験(ラットにおける出生前及び出生後の発生並びに母体の機能に関する試験)の12mg/kg/日(60mg/日でのヒトの血漿中曝露量の11.1倍)以上で出生児の腎臓にキサンチンと推定される結晶沈着あるいは結石、48mg/kg/日(60mg/日でのヒトの血漿中曝露量の39.3倍)で離乳率の低下、体重低値などの発育抑制、甲状腺の大型化及び甲状腺重量増加の傾向が認められている1)。]
小児等への投与
低出生体重児、新生児、乳児、幼児又は小児に対する安全性は確立していない。[使用経験がない。]
適用上の注意
薬剤交付時
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。[PTPシートの誤飲により、硬い鋭角部が食道粘膜に刺入し、更には穿孔を起こして縦隔洞炎等の重篤な合併症を誘発することが報告されている。]
その他の注意
げっ歯類を用いた104週間投与によるがん原性試験において、最高用量群(ラット24mg/kg/日〔60mg/日でのヒトの血漿中曝露量の約25(雄)及び26(雌)倍〕、マウス18.75mg/kg/日〔60mg/日でのヒトの血漿中曝露量の約4(雄)及び12(雌)倍〕)の膀胱にキサンチンと推定される結晶沈着・結石が認められ、マウスの18.75mg/kg/日(雌)及びラットの24mg/kg/日(雄)に膀胱腫瘍(移行上皮乳頭腫及び移行上皮癌)の発生頻度の増加が認められた。マウスでは膀胱にキサンチン結晶・結石が生成しない条件下で、膀胱移行上皮の過形成は認められなかった。げっ歯類では、結晶・結石などによる機械的刺激が長時間持続することにより、膀胱粘膜の腫瘍性変化が誘発されるとの報告がある。また、臨床試験において、キサンチン結晶・結石を疑わせる尿沈渣所見はなかった2)。
薬物動態
1.
血漿中濃度
(1)
単回投与
健康成人男性30例に、フェブキソスタットとして10、20、40及び80mgを絶食下で単回経口投与したとき、血漿中フェブキソスタットの濃度推移及び薬物動態パラメータは以下のとおりである 3),4)。  (表1参照) (2)
反復投与
健康成人男性6例に、フェブキソスタットとして40mgを朝食後に1日1回7日間反復経口投与したとき、血漿中フェブキソスタット濃度は投与開始後3日で定常状態に達し、反復投与による蓄積性は認められなかった5)。
(表2参照)
(3)
反復投与(高尿酸血症患者)
高尿酸血症患者10例にフェブキソスタット10mg/日で2週間、20mg/日を4週間1日1回朝食後に投与したとき、投与開始後6週における薬物動態パラメータは以下のとおりであった6)。
(表3参照)
(4)
食事の影響
健康成人16例に、フェブキソスタット40mgを食後に単回経口投与したとき、空腹時投与に比べて、Cmax及びAUCinfはそれぞれ28及び18%低下した3)。
(表4参照)
2.
特殊集団における薬物動態
(1)
腎機能低下患者
軽度(5例)及び中等度(7例)の腎機能低下患者にフェブキソスタット20mgを1日1回朝食後に7日間反復経口投与したとき、投与後7日における軽度腎機能低下群のフェブキソスタットのCmaxは腎機能正常群(9例)と変わらなかったが、AUC0,24hrは腎機能正常群に比較して53%増加した。中等度腎機能低下群のCmax及びAUC0,24hrは腎機能正常群に比較して、それぞれ26及び68%増加した7)。
軽度(6例)、中等度(7例)及び重度(7例)の腎機能低下患者にフェブキソスタット80mgを1日1回朝食前に7日間反復経口投与したとき、投与後7日におけるフェブキソスタットのCmax及びAUC0,24hrは、腎機能正常群(11例)に比較して軽度、中等度、重度腎機能低下群でそれぞれ41及び48%、2及び48%、4及び76%上昇した(外国人のデータ)8)。
(2)
肝機能低下患者
軽度(8例)及び中等度(8例)の肝機能低下患者(Child-Pugh A、B)にフェブキソスタット80mgを1日1回朝食前に7日間反復経口投与したとき、軽度肝機能低下群の投与後7日におけるフェブキソスタットのCmax及びAUC0,24hrは、肝機能正常群(11例)と比較してそれぞれ24及び30%上昇した。また、中等度肝機能低下群のCmax及びAUC0,24hrはそれぞれ53及び55%上昇した(外国人のデータ)9)。
(3)
高齢者及び性別の影響
高齢者(65歳以上、24例)と若年者(18~40歳、24例)にフェブキソスタット80mgを1日1回朝食前に7日間反復経口投与したとき、投与後7日における高齢者のCmax及びAUC0,24hrは若年者に対してそれぞれ1%低下及び12%上昇した。
また、女性被験者群(24例)のCmax及びAUC0,24hrは男性被験者群(24例)に比較してそれぞれ24及び12%高かった(外国人のデータ)10)。
(注)本剤の承認された用法・用量における最大投与量は60mg/日である。
3.
蛋白結合率
フェブキソスタット(0.4~10 μ g/mL添加時)のヒト血漿蛋白結合率は97.8~99.0%であり、主な結合蛋白はアルブミンであった(in vitro 試験)11)。
4.
代謝・排泄
(1)
フェブキソスタットの主な代謝経路はグルクロン酸抱合反応であった。また、その他に複数の酸化代謝物、それらの硫酸抱合体及びグルクロン酸抱合体に代謝された12)。
フェブキソスタットのCYP1A2、CYP2B6、CYP2C9、CYP2C19、及びCYP3A4/5に対する阻害は認められなかった。一方、フェブキソスタットのCYP2C8及びCYP2D6に対するKi値はそれぞれ20及び40μmol/Lであった(ヒト肝ミクロソームを用いたin vitro 試験)11),13)。
フェブキソスタットはCYP1A1/2、CYP2B6、CYP2C9、CYP2C19、CYP2D6、CYP2E1及びCYP3A4/5を誘導しなかった(ヒト初代肝細胞を用いたin vitro 試験)13)。
(2)
健康成人男性24例にフェブキソスタットとして10、20、40mgを絶食下単回経口投与したとき、投与後24及び96時間までの投与量に対するフェブキソスタットの尿中排泄率はそれぞれ2.1~3.8%及び2.2~3.9%であった。また、投与後24及び96時間までの投与量に対するフェブキソスタットのグルクロン酸抱合体の尿中排泄率はそれぞれ46.7~49.7%及び49.0~51.6%であった3)。
(3)
健康成人男性6例に14C-フェブキソスタットとして80mgを含有する液剤を、絶食下単回経口投与したとき、投与後4時間までの血漿中総放射能に対するフェブキソスタット及びそのグルクロン酸抱合体の割合はそれぞれ83.8~95.8%及び2.3~6.8%であった。投与後48時間までのフェブキソスタットの尿中排泄率(投与量に対する割合、以下同様)は1.1~3.5%、投与後120時間までの糞中排泄率は7.8~15.8%であった。また、代謝物を含めた総放射能の投与後216時間までの尿及び糞中排泄率はそれぞれ49.1及び44.9%であった(外国人のデータ)12)。
(注)本剤の承認された用法・用量における最大投与量は60mg/日である。
5.
薬物相互作用
(1)
制酸剤の影響
健康成人24例に制酸剤(5mL中に水酸化マグネシウム200mg及び水酸化アルミニウム225mgを含有する配合剤)を単回経口投与後にフェブキソスタット80mgを単回経口投与したとき、フェブキソスタットのCmax及びAUCinfはそれぞれ32及び15%低下した(外国人のデータ)14)。
(2)
コルヒチンの影響及びコルヒチンへの影響
健康成人22例にフェブキソスタット40mgを1日1回7日間反復経口投与し、更に4~7日目にコルヒチンを1.2mg/日で1日2回反復経口投与したとき、フェブキソスタットのCmax及びAUC0,24hrはそれぞれ12及び7%上昇した(外国人のデータ)15)。
健康成人26例にフェブキソスタット120mgを1日1回及びコルヒチンを1.2mg/日で1日2回14日間反復経口投与したとき、コルヒチンの朝食前投与後もしくは夕食後投与後のCmaxはそれぞれ12%低下及び2%上昇した。また、AUC0,24hrは3%低下した15)。
(3)
インドメタシンの影響及びインドメタシンへの影響
健康成人26例にフェブキソスタット80mgを1日1回及びインドメタシン100mg/日で1日2回5日間反復経口投与したとき、フェブキソスタットのCmaxは7%低下し、AUC0,24hrは2%上昇した。また、インドメタシンのCmax及びAUC0,24hrの低下は2%以内であった(外国人のデータ)16)。
(4)
ナプロキセンの影響及びナプロキセンへの影響
健康成人25例にフェブキソスタット80mgを1日1回及びナプロキセン1000mg/日を1日2回7日間反復経口投与したとき、ナプロキセンの併用により、フェブキソスタットのCmax及びAUC0,24hrはそれぞれ28及び40%上昇した。一方、フェブキソスタットの併用によるナプロキセンのCmaxの上昇及びAUC0,24hrの低下は1%以内であった(外国人のデータ)16)。
(5)
デシプラミンへの影響
健康成人18例にフェブキソスタット120mgを1日1回9日間反復経口投与し、投与開始後6日にデシプラミン(国内未承認)25mgを単回経口投与したとき、デシプラミンのCmax及びAUCinfはそれぞれ16及び22%上昇した(外国人のデータ)17)。
(6)
ワルファリンナトリウムへの影響
健康成人13例にフェブキソスタット120mgとワルファリンナトリウム(国内未承認)(用量はINRを基準に設定)を1日1回14日間反復経口投与したとき、R-及びS-ワルファリンのCmax及びAUC0,24hrの上昇は5%以内であった。また、ワルファリンの薬力学の変化(INRmaxの上昇、INRmean,24hの低下及び第VII因子活性平均値の上昇)は7%以内であった(外国人のデータ)18)。
健康成人27例にフェブキソスタット80mgとワルファリンナトリウム(国内未承認)(用量はINRを基準に設定)を1日1回14日間反復経口投与したとき、R-ワルファリンのCmax及びAUC0,24hrの低下は2%以内であった。また、S-ワルファリンのCmaxの低下及びAUC0,24hrの上昇は1%以内であった。また、ワルファリンの薬力学の変化(INRmaxとINRmean,24hの低下及び第VII因子活性平均値の上昇)は4%以内であった(外国人のデータ)18)。
(7)
ヒドロクロロチアジドの影響
健康成人33例にフェブキソスタット80mgとヒドロクロロチアジド50mgを単回経口投与したとき、フェブキソスタットのCmax及びAUCinfの上昇は4%以内であった(外国人のデータ)19)。
(8)
テオフィリンへの影響
健康成人23例にフェブキソスタット80mgを1日1回7日間反復経口投与し、投与開始後5日にテオフィリン400mgを単回経口投与したとき、テオフィリンのCmax及びAUCinfの上昇は5% 以内であった(外国人のデータ)20)。
(9)
ロシグリタゾンへの影響
健康成人36例にフェブキソスタット120mgを1日1回9日間反復経口投与し、投与開始後5日にロシグリタゾン(国内未承認)4mgを単回経口投与したとき、ロシグリタゾンのCmaxの低下及びAUCinfの上昇は6%以内であった21)。
(注)本剤の承認された用法・用量における最大投与量は60mg/日である。
表1
| 用量 |
薬物動態パラメータ
AUCinf
(ng・hr/mL) |
薬物動態パラメータ
Cmax
(ng/mL) |
薬物動態パラメータ
t1/2
(hr) |
薬物動態パラメータ
tmax
(hr) |
| 10mg(N=8) |
1537.0±430.9 |
496.2±166.0 |
6.2±0.9 |
1.4±1.1 |
| 20mg(N=8) |
3296.2±751.9 |
1088.3±178.9 |
6.2±1.1 |
1.3±0.5 |
| 40mg(N=8) |
7085.2±1341.2 |
2270.3±866.7 |
7.3±1.8 |
1.2±0.8 |
| 80mg(N=6) |
13300.5±3032.3 |
3765.3±1008.3 |
6.9±1.8 |
1.9±1.0 |
(平均値±標準偏差)
表2
| 用量 |
観察日 |
Cmax
(ng/mL) |
tmax
(hr) |
AUC0,24hr
(ng・hr/mL) |
t1/2
(hr) |
| 40mg/日(N=6) |
1日目 |
1019.1±343.2 |
1.8±0.8 |
3658.5±625.6 |
6.3±1.6 |
| 40mg/日(N=6) |
7日目 |
1299.8±312.6 |
1.5±0.3 |
4442.1±729.5 |
8.8±2.2 |
(平均値±標準偏差)
表3
| 投与群 |
Cmax
(ng/mL) |
tmax
(hr) |
AUC0,24hr
(ng・hr/mL) |
t1/2
(hr) |
| 20mg(N=10) |
541.8±227.8 |
2.2±1.6 |
2092.3±463.2 |
8.2±2.4 |
(平均値±標準偏差)
表4
| 投与群 |
Cmax
(ng/mL) |
tmax
(hr) |
AUCinf
(ng・hr/mL) |
t1/2
(hr) |
| 絶食下投与(N=16) |
2049.1±782.3 |
1.2±0.8 |
6538.3±1263.0 |
6.8±1.7 |
| 食後投与(N=16) |
1456.0±514.8 |
1.8±1.0 |
5321.6±910.4 |
6.3±1.5 |
(平均値±標準偏差)
臨床成績
1.
痛風を含む高尿酸血症患者202例を対象としたプラセボ対照無作為化二重盲検用量反応比較試験を行った。本剤10mg/日から投与を開始し、各群の固定維持用量(20、40、60又は80mg/日)まで用量を段階的に増量した。増量のタイミングは投与開始後2、6及び10週とし、これ以降16週まで用量を維持した。投与開始後16週時(各群の維持用量まで用量を段階的に増量した期間を含む)に、血清尿酸値が6.0mg/dL以下に到達した患者の割合(達成率)及び各投与期間での痛風関節炎の発現割合は、下表のとおりであった22)。
(表5参照)
(表6参照)
(注)本剤の承認された用法・用量における最大投与量は60mg/日である。
2.
痛風を含む高尿酸血症患者244例を対象としたアロプリノール対照無作為化二重盲検比較試験を行った。本剤10mg/日又はアロプリノール100mg/日を12日間投与し、その後、それぞれ40mg/日又は200mg/日に増量し44日間投与した。投与開始後8週の血清尿酸値変化率(主要評価項目)において、本剤40mg/日群のアロプリノール200mg/日に対する非劣性が示された(P<0.001:非劣性マージンは5%)。また、投与開始後8週の血清尿酸値6.0mg/dL以下達成率(副次評価項目)は、本剤40mg/日群82.0%、アロプリノール200mg/日群70.0%であった。各投与期間での痛風関節炎の発現割合は下表のとおりであった23)。
(表7参照)
(表8参照)
3.
血清尿酸値9.0mg/dL以上の痛風を含む高尿酸血症患者171例を対象とした長期投与試験を実施した。10mg/日から投与を開始し、投与開始後3週目に20mg/日、投与開始後7週目に40mg/日に増量した。投与開始後10週目の血清尿酸値が6.0mg/dLを超えていた場合は投与開始後15週目より60mg/日に増量し、血清尿酸値が6.0mg/dL以下の場合は40mg/日を維持した。投与開始後18週、26週、52週で血清尿酸値が6.0mg/dL以下に到達した患者の割合(達成率)は、40mg投与群では、それぞれ93.5、91.5、86.4%、また、60mg投与群では、それぞれ74.4、71.4、87.5%であった24)。
表5 投与開始後16週の血清尿酸値6.0mg/dL以下達成率
| 投与群 |
血清尿酸値
6.0mg/dL以下
達成率 |
プラセボ群との差 |
95%信頼区間
(%) |
| プラセボ(38例) |
2.6% |
- |
- |
| 20mg/日(43例) |
46.5% |
43.9% |
28.1~59.6 |
| 40mg/日(41例) |
82.9% |
80.3% |
67.7~92.9 |
| 60mg/日(36例) |
83.3% |
80.7% |
67.5~93.9 |
| 80mg/日(41例) |
87.8% |
85.2% |
73.9~96.4 |
表6 痛風関節炎の発現割合
| 投与群 |
0~2週
以下 |
2週超
6週以下 |
6週超
10週以下 |
10週超
16週以下 |
| プラセボ(38例) |
0.0% |
5.3% |
2.6% |
2.7% |
| 20mg/日群(43例) |
0.6%(10mg/日) |
2.5%(20mg/日) |
4.9%(20mg/日) |
2.4%(20mg/日) |
| 40mg/日群(41例) |
0.6%(10mg/日) |
2.5%(20mg/日) |
3.4%(40mg/日) |
7.5%(40mg/日) |
| 60mg/日群(36例) |
0.6%(10mg/日) |
2.5%(20mg/日) |
3.4%(40mg/日) |
8.8%(60mg/日) |
| 80mg/日群(41例) |
0.6%(10mg/日) |
2.5%(20mg/日) |
3.4%(40mg/日) |
17.9%(80mg/日) |
()内は当該時期の本剤の用量
表7 投与開始後8週の血清尿酸値変化率(%)
| 投与群 |
血清尿酸初期値
(mg/dL)
平均(標準偏差) |
血清尿酸値
変化率a)
(%)
平均(標準偏差) |
変化率の群間差
[95%信頼区間]
(%) |
共分散
分析 |
| アロプリノール200mg/日(120例) |
8.89(1.24) |
-35.2(14.7) |
-6.24[-9.65,-2.84] |
P<0.001※ |
| 本剤40mg/日(122例) |
8.83(1.32) |
-41.5(12.1) |
-6.24[-9.65,-2.84] |
P<0.001※ |
a)血清尿酸初期値(投与開始前の血清尿酸値)に対する投与開始後8週の血清尿酸値の変化率 ※非劣性検定におけるP値
表8 痛風関節炎の発現割合
| 投与群 |
0~12日以下 |
12日超
6週以下 |
6週超
8週以下 |
| アロプリノール200mg/日(121例) |
1.7%(100mg/日) |
3.3%(200mg/日) |
0.9%(200mg/日) |
| 本剤40mg/日(122例) |
1.6%(10mg/日) |
5.7%(40mg/日) |
3.3%(40mg/日) |
()内は当該時期のアロプリノール又は本剤の用量
薬効薬理
1.
作用機序
(1)
フェブキソスタットは、尿酸生成を掌るキサンチンオキシダーゼの酸化型(Ki値:0.6nmol/L)、還元型(Ki値:3.1nmol/L)をいずれも阻害することにより、尿酸生成を抑制する(in vitro 試験)25)。
(2)
フェブキソスタットは、他の主要なプリン・ピリミジン代謝酵素の活性に影響を及ぼさず、キサンチンオキシダーゼを選択的に阻害する(in vitro 試験)25)。
2.
薬理作用
(1)
血中尿酸低下作用
ラット(正常、高尿酸血症モデル)でフェブキソスタットは経口投与により、血中尿酸値を低下させた26)。
(2)
尿中尿酸低下作用
ラットでフェブキソスタットは経口投与により、尿中尿酸値を低下させた26)。
有効成分に関する理化学的知見
一般名
フェブキソスタット(Febuxostat)
化学名
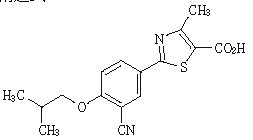
2-[3-cyano-4-(2-methylpropoxy)phenyl]-4-methylthiazole-5-carboxylic acid
化学構造式
分子式
C16H16N2O3S
分子量
316.37
融点
約209℃(分解)
性状
白色の粉末。N ,N -ジメチルホルムアミドに溶けやすく、ジメチルスルホキシドにやや溶けやすく、エタノール(99.5)にやや溶けにくく、メタノール及びアセトニトリルに溶けにくく、水にはほとんど溶けない。
包装

フェブリク錠10mg
PTP:140錠(14錠×10)
フェブリク錠20mg
PTP:70錠(14錠×5)、500錠(10錠×50)
瓶:500錠(プラスチック製瓶入り)
フェブリク錠40mg
PTP:140錠(14錠×10)
主要文献及び文献請求先
主要文献
1)
社内報告:生殖発生毒性(ラット), 2010
2)
社内報告:がん原性(マウス、ラット), 2010
3)
社内報告:血漿中濃度及び排泄(健康成人、単回), 2010
4)
社内報告:血漿中濃度(健康成人、単回), 2010
5)
社内報告:血漿中濃度(健康成人、反復), 2010
6)
社内報告:血漿中濃度(高尿酸血症患者、反復), 2010
7)
社内報告:血漿中濃度(腎機能低下患者、反復), 2010
8)
Mayer, M. D. et al.:Am. J. Ther., 12(1)22, 2005
9)
Khosravan, R. et al.:J. Clin. Pharmacol., 46(1)88, 2006
10)
Khosravan, R. et al.:J. Clin. Pharmacol., 48(9)1014, 2008
11)
Mukoyoshi, M. et al.:Xenobiotica., 38(5)496, 2008
12)
Grabowski, B. A. et al.:J. Clin. Pharmacol., 51(2)189, 2011
13)
社内報告:代謝(ヒト), 2010
14)
Khosravan, R. et al.:Br. J. Clin. Pharmacol., 65(3)355, 2008
15)
社内報告:薬物相互作用(コルヒチン), 2010
16)
Khosravan, R. et al.:J. Clin. Pharmacol., 46(8)855, 2006
17)
社内報告:薬物相互作用(デシプラミン), 2010
18)
社内報告:薬物相互作用(ワルファリン), 2010
19)
Grabowski, B. A. et al.:Br. J. Clin. Pharmacol., 70(1)57, 2010
20)
社内報告:薬物相互作用(テオフィリン), 2010
21)
社内報告:薬物相互作用(ロシグリタゾン), 2010
22)
社内報告:プラセボ対照無作為化二重盲検用量反応比較試験(痛風を含む高尿酸血症患者), 2010
23)
社内報告:アロプリノール対照無作為化二重盲検比較試験(痛風を含む高尿酸血症患者), 2010
24)
社内報告:長期投与試験(痛風を含む高尿酸血症患者), 2010
25)
Takano, Y. et al.:Life Sci., 76(16)1835, 2005
26)
社内報告:血中及び尿中尿酸低下作用(ラット), 2010
文献請求先
主要文献に記載の社内報告につきましても下記にご請求ください。
帝人ファーマ株式会社 学術情報部
〒100-8585 東京都千代田区霞が関3丁目2番1号
電話 03-3506-4053
長期投与医薬品に関する情報
本剤は新医薬品であるため、厚生労働省告示第97号(平成20年3月19日付)に基づき、平成24年3月末日までは、投薬は1回14日分を限度とされています。
製造販売業者等の氏名又は名称及び住所
製造販売元
帝人ファーマ株式会社
東京都千代田区霞が関3丁目2番1号
|