|
英文药名:XGEVA(denosumab injection)
中文药名:地诺单抗(狄迪诺塞麦注射剂)
生产商:安进(Amgen)
药品介绍
Xgeva(denosumab,狄迪诺塞麦)是一种RANK配体(RANKL)抑制剂,由安进公司(Amgen)生产,2010年11月18日批准用于防止已扩散到骨的癌症所造成的骨相关事件,2012年4月份FDA将会决定是否批准用于预防前列腺癌骨转移
作用机理:
地诺单抗与现已获准的减少肿瘤骨骼并发症的药物作用机制不同。它是一种特异性靶向核因子-κB受体活化因子配体(RANKL)的完全人源化单克隆抗体(IgG2单抗),阻止RANKL和其受体物质结合,抑制破骨细胞活化和发展,减少骨吸收,增加骨密度。早前地诺单抗(商品名:Prolia)被用来治疗有较高骨折风险的绝经后妇女骨质疏松症,具有较好的安全性和有效性。
适应证和用途
Xgeva是一种RANK配体(RANKL)抑制剂适用于:
(1)在有实体瘤骨转移患者中骨骼相关事件的预防
使用的重要限制:Xgeva不适用于在多发性骨髓瘤患者中为预防骨骼相关事件
剂量和给药方法
(1)在上臂,上大腿,或腹部皮下注射给予120 mg每4周1次
(2)当需要治疗或预防低钙血症给予钙和维生素D
剂型和规格
(1)120mg/1.7mL(70mg/mL)单次使用小瓶
禁忌证
(1)无
警告和注意事项
(1)接受Xgeva患者中可能发生低钙血症,严重低钙血症。开始Xgeva前纠正低钙血
症。监视钙水平和用钙和维生素D适当补充所有患者。
(2)接受Xgeva患者中可能发生颚骨坏死。开始Xgeva前进行口腔检查。监视症状。用Xgeva治疗期间避免侵害性牙科手术。
不良反应
(1)接受Xgeva患者中最常见不良反应(每例-患者发生率大于或等于25%)是疲劳/虚弱, 低磷酸盐血症,和恶心。
特殊人群中使用
(1)妊娠:根据动物资料,可能引起胎儿损害。可供利用妊娠监察计划。
(2)哺乳母亲:乳腺发育和哺乳可能受损。终止药物或哺乳。
(3)儿童患者:未确定安慰性和有效性。
(4)肾受损:患者with肌酐清除率低于30mL/min或接受透析是低钙血症风险。适当补充钙和维生素D。
保存:
贮藏在冰箱内在2至8℃(36至46℉)在原始盒内。不要冻结,一旦从冰箱取出,不要暴露在温度25℃/77℉以上或光线,必须在14天内使用。
建议:
目前在预防骨相关事件的药物中,还没有足够的证据显示地诺单抗明显优于其它任一种药物,特别在延长患者生存期方面。因此,选择时,要综合考虑患者体质状况、经济状况、药物毒副反应、个人喜好等情况,以给患者带来最大的益处为准则来选用。
临床研究:
癌症骨转移是患者疼痛重要原因,影响患者的生活质量。三项随机-双盲的临床研究证实了地诺单抗的安全性和疗效,总共有5723名患者参与研究。这些研究在乳腺癌、前列腺癌以及多种其它癌症患者身上对地诺单抗与唑来膦酸作了比较。
这些研究旨在测定由于癌症,患者最终出现骨折或脊髓压迫,或为控制疼痛需要进行放射或手术治疗间隔的时间。
在前列腺癌患者中,地诺单抗延迟SREs优于唑来膦酸。前列腺癌患者中SREs延迟时间中位值21个月,唑来膦酸为17个月。
在乳腺癌患者中,唑来膦酸使SREs延迟的中位时间为26个月,而地诺单抗没有达到这一水平。
在其它实体瘤患者中,地诺单抗和唑来膦酸使SREs延迟的中位时间不相上下。受试者主要的实体瘤为非小细胞肺癌、多发性骨髓瘤、肾癌以及小细胞肺癌。
-----------------------------------------------
Xgeva(denosumab)Injection
1. Name of the medicinal product
XGEVA®▼ 120 mg solution for injection
2. Qualitative and quantitative composition
Each vial contains 120 mg of denosumab in 1.7 ml of solution (70 mg/ml).
Denosumab is a human monoclonal IgG2 antibody produced in a mammalian cell line (CHO) by recombinant DNA technology.
Excipient with known effects:
Each 1.7 ml of solution contains 78 mg sorbitol (E420).
For the full list of excipients, see section 6.1.
3. Pharmaceutical form
Solution for injection (injection).
Clear, colourless to slightly yellow solution and may contain trace amounts of translucent to white proteinaceous particles.
4. Clinical particulars
4.1 Therapeutic indications
Prevention of skeletal related events (pathological fracture, radiation to bone, spinal cord compression or surgery to bone) in adults with bone metastases from solid tumours.
4.2 Posology and method of administration
Posology
The recommended dose of XGEVA is 120 mg administered as a single subcutaneous injection once every 4 weeks into the thigh, abdomen or upper arm.
Supplementation of at least 500 mg calcium and 400 IU vitamin D daily is required in all patients, unless hypercalcaemia is present (see section 4.4).
Patients with renal impairment
No dose adjustment is required in patients with renal impairment (see section 5.2). Experience in patients on dialysis or with severe renal impairment (creatinine clearance < 30 ml/min) is limited (see section 4.4 for recommendations relating to monitoring of calcium).
Patients with hepatic impairment
The safety and efficacy of denosumab have not been studied in patients with hepatic impairment (see section 5.2).
Elderly patients (age ≥ 65)
No dose adjustment is required in elderly patients (see section 5.2).
Paediatric population
XGEVA is not recommended in paediatric patients (age < 18) as the safety and efficacy of XGEVA in these patients have not been established. Inhibition of RANK/RANK ligand (RANKL) in animal studies has been coupled to inhibition of bone growth and lack of tooth eruption, and these changes were partially reversible upon cessation of RANKL inhibition (see section 5.3).
Method of administration
For subcutaneous use.
XGEVA should be administered under the responsibility of a healthcare professional.
The instructions for use, handling and disposal are given in section 6.6.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Severe, untreated hypocalcaemia (see section 4.4).
4.4 Special warnings and precautions for use
Calcium and Vitamin D supplementation
Supplementation with calcium and vitamin D is required in all patients unless hypercalcaemia is present (see section 4.2).
Hypocalcaemia
Pre-existing hypocalcaemia must be corrected prior to initiating therapy with XGEVA. Hypocalcaemia can occur at any time during therapy with XGEVA and most commonly occurs within the first 6 months of dosing. Patients with severe renal impairment (creatinine clearance < 30 ml/min) or receiving dialysis are at greater risk of developing hypocalcaemia. Monitoring of calcium levels in these patients is recommended. If hypocalcaemia occurs while receiving XGEVA, additional calcium supplementation may be necessary.
In the post marketing setting, severe symptomatic hypocalcaemia (including fatal cases) has been reported (see section 4.8).
Osteonecrosis of the jaw
Osteonecrosis of the jaw (ONJ) has occurred in patients treated with XGEVA . In clinical trials, the incidence of ONJ was higher with longer duration of exposure (see section 4.8).
Patients who developed ONJ in clinical studies generally had known risk factors for ONJ, including invasive dental procedures (e.g., tooth extraction, dental implants, oral surgery), poor oral hygiene or other pre-existing dental disease, advanced malignancies, infections, or concomitant therapies (e.g., chemotherapy, corticosteroids, angiogenesis inhibitors, radiotherapy to the head and neck). A dental examination with appropriate preventive dentistry should be considered prior to treatment with XGEVA in patients with active dental and jaw conditions (as listed above). While on treatment, patients should avoid invasive dental procedures if possible.
Good oral hygiene practices should be maintained during treatment with XGEVA. Patients who are suspected of having or who develop ONJ while on XGEVA therapy should receive care by a dentist or oral surgeon. In these patients, extensive dental surgery to treat ONJ may exacerbate the condition.
An individual risk/benefit evaluation should be done for each patient before prescribing XGEVA in patients with unavoidable risk factors for ONJ; and in patients who have developed ONJ during treatment with XGEVA.
Atypical fractures of the femur
Atypical femoral fractures have been reported in patients receiving XGEVA (see section 4.8). Atypical femoral fractures may occur with little or no trauma in the subtrochanteric and diaphyseal regions of the femur. Specific radiographic findings characterise these events. Atypical femoral fractures have also been reported in patients with certain comorbid conditions (e.g. vitamin D deficiency, rheumatoid arthiritis, hypophosphatasia) and with use of certain pharmaceutical agents (e.g.bisphosphonates, glucocorticoids, proton pump inhibitors). These events have also occurred without antiresorptive therapy. Similar fractures reported in association with bisphosphonates are often bilateral; therefore the contralateral femur should be examined in denosumab-treated patients who have sustained a femoral shaft fracture. Discontinuation of XGEVA therapy in patients suspected to have an atypical femur fracture should be considered pending evaluation of the patient based on an individual benefit risk assessment. During XGEVA treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Patients presenting with such symptoms should be evaluated for an incomplete femoral fracture.
Others
Patients being treated with XGEVA should not be treated concomitantly with other denosumab containing medicinal products (for osteoporosis indications).
Patients being treated with XGEVA should not be treated concomitantly with bisphosphonates.
Warnings for excipients
Patients with rare hereditary problems of fructose intolerance should not use XGEVA.
4.5 Interaction with other medicinal products and other forms of interaction
No interaction studies have been performed.
In clinical trials, XGEVA has been administered in combination with standard anti-cancer treatment and in subjects previously receiving bisphosphonates. There were no clinically-relevant alterations in trough serum concentration and pharmacodynamics of denosumab (creatinine adjusted urinary N-telopeptide, uNTx/Cr) by concomitant chemotherapy and/or hormone therapy or by previous intravenous bisphosphonate exposure.
4.6 Fertility, pregnancy and lactation
Pregnancy
There are no adequate data from the use of XGEVA in pregnant women. Reproductive toxicity was shown in a study of cynomolgus monkeys, dosed throughout pregnancy with denosumab at AUC exposures 12-fold higher than the human dose (see section 5.3).
XGEVA is not recommended for use in pregnant women and women of childbearing potential not using contraception.
Women who become pregnant during XGEVA treatment are encouraged to enrol in Amgen's Pregnancy Surveillance Programme. Contact details are provided in section 6 of the Package Leaflet.
Breast-feeding
It is unknown whether denosumab is excreted in human milk. Knockout mouse studies suggest absence of RANKL during pregnancy may interfere with maturation of the mammary gland leading to impaired lactation post-partum (see section 5.3). A decision on whether to abstain from breast-feeding or to abstain from therapy with XGEVA should be made, taking into account the benefit of breast-feeding to the newborn/infant and the benefit of XGEVA therapy to the woman.
Women who are nursing during XGEVA treatment are encouraged to enrol in Amgen's Lactation Surveillance Programme. Contact details are provided in section 6 of the Package Leaflet.
Fertility
No data are available on the effect of denosumab on human fertility. Animal studies do not indicate direct or indirect harmful effects with respect to fertility (see section 5.3).
4.7 Effects on ability to drive and use machines
XGEVA has no or negligible influence on the ability to drive and use machines.
4.8 Undesirable effects
Summary of the safety profile
The safety of XGEVA was evaluated in:
• 5,931 patients with advanced malignancies involving bone and is derived from active-controlled, clinical trials examining the efficacy and safety of XGEVA versus zoledronic acid in preventing the occurrence of skeletal related events.
The adverse reactions are presented in table 1.
Tabulated list of adverse reactions
The following convention has been used for the classification of the adverse reactions reported in three phase III and one phase II clinical studies (see table 1): very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000) and very rare (< 1/10,000). Within each frequency grouping and system organ class, adverse reactions are presented in order of decreasing seriousness.
Table 1 Adverse reactions reported in patients with advanced malignancies involving bone
|
MedDRA system organ class |
Frequency category |
Adverse reactions |
|
Immune system disorder |
Rare |
Drug hypersensitivity1 |
|
Rare |
Anaphylactic reaction1 |
|
Metabolism and nutrition disorders |
Common |
Hypocalcaemia1 |
|
Common |
Hypophosphataemia |
|
Respiratory, thoracic and mediastinal disorders |
Very common |
Dyspnoea |
|
Gastrointestinal disorders |
Very common |
Diarrhoea |
|
Common |
Tooth extraction |
|
Skin and subcutaneous tissues disorders |
Common |
Hyperhidrosis |
|
Musculoskeletal and connective tissue disorders |
Common |
Osteonecrosis of the jaw1 |
|
Rare |
Atypical femoral fracture1 |
1 See section Description of selected adverse reactions
Description of selected adverse reactions
Hypocalcaemia
In three phase III active-controlled clinical trials in patients with advanced malignancies involving bone, hypocalcaemia was reported in 9.6% of patients treated with XGEVA and 5.0% of patients treated with zoledronic acid.
A grade 3 decrease in serum calcium levels was experienced in 2.5% of patients treated with XGEVA and 1.2% of patients treated with zoledronic acid. A grade 4 decrease in serum calcium levels was experienced in 0.6% of patients treated with XGEVA and 0.2% of patients treated with zoledronic acid (see section 4.4).
Symptoms of hypocalcaemia in clinical studies included paresthesias or muscle stiffness, twitching, spasms and muscle cramps.
In the post-marketing setting, severe symptomatic hypocalcaemia (including fatal cases) has been reported.
Osteonecrosis of the jaw (ONJ)
In the primary treatment phases of three phase III active-controlled clinical trials in patients with advanced malignancies involving bone, ONJ was confirmed in 1.8% of patients treated with XGEVA (median exposure of 12.0 months; range 0.1 – 40.5) and 1.3% of patients treated with zoledronic acid. Clinical characteristics of these cases were similar between treatment groups. Among subjects with confirmed ONJ, most (81% in both treatment groups) had a history of tooth extraction, poor oral hygiene, and/or use of a dental appliance. In addition most subjects were receiving or had received chemotherapy. The trials in patients with breast or prostate cancer included an XGEVA extension treatment phase (median overall exposure of 14.9 months; range 0.1 – 67.2). The patient-year adjusted incidence of confirmed ONJ was 1.1% during the first year of treatment and 4.1% thereafter. The median time to ONJ was 20.6 months (range: 4 - 53). Patients with certain identified risk factors for ONJ were excluded from participation in the pivotal studies.
Drug related hypersensitivity reactions
In the post-marketing setting, events of hypersensitivity, including rare events of anaphylactic reactions, have been reported in patients receiving XGEVA.
Atypical fractures of the femur
In the clinical trial program, atypical femoral fractures were reported rarely in patients treated with denosumab (see section 4.4).
Other special populations
In a clinical study of patients without advanced cancer with severe renal impairment (creatinine clearance < 30 ml/min) or receiving dialysis, there was a greater risk of developing hypocalcaemia in the absence of calcium supplementation.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via:
United Kingdom
Yellow Card Scheme
Tel: Freephone 0808 100 3352
Website: www.mhra.gov.uk/yellowcard
Ireland
Pharmacovigilance Section
Irish Medicines Board
Kevin O'Malley House
Earlsfort Centre
Earlsfort Terrace
IRL - Dublin 2
Tel: +353 1 6764971
Fax: +353 1 6762517
Website: www.imb.ie
e-mail: imbpharmacovigilance@imb.ie
4.9 Overdose
There is no experience with overdose in clinical studies. XGEVA has been administered in clinical studies using doses up to 180 mg every 4 weeks and 120 mg weekly for 3 weeks.
5. Pharmacological properties
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: Drugs for the treatment of bone diseases – other drugs affecting bone structure and mineralisation, ATC code: M05BX04
Mechanism of action
RANKL exists as a transmembrane or soluble protein. RANKL is essential for the formation, function and survival of osteoclasts, the sole cell type responsible for bone resorption. Increased osteoclast activity, stimulated by RANKL, is a key mediator of bone destruction in metastatic bone disease and multiple myeloma. Denosumab is a human monoclonal antibody (IgG2) that targets and binds with high affinity and specificity to RANKL, preventing the RANKL/RANK interaction from occurring and resulting in reduced osteoclast numbers and function, thereby decreasing bone resorption and cancer-induced bone destruction.
Pharmacodynamic effects
In phase II clinical studies of patients with advanced malignancies involving bone, subcutaneous (SC) dosing of XGEVA administered either every 4 weeks or every 12 weeks resulted in a rapid reduction in markers of bone resorption (uNTx/Cr, serum CTx), with median reductions of approximately 80% for uNTx/Cr occurring within 1 week regardless of prior bisphosphonate therapy or baseline uNTx/Cr level. In the phase III clinical trials, median reductions of approximately 80% were maintained in uNTx/Cr after 3 months of treatment in 2075 XGEVA-treated advanced cancer patients naïve to IV-bisphosphonate.
Immunogenicity
In clinical studies, neutralising antibodies have not been observed for XGEVA. Using a sensitive immunoassay < 1% of patients treated with denosumab for up to 3 years tested positive for non neutralising binding antibodies with no evidence of altered pharmacokinetics, toxicity, or clinical response.
Clinical efficacy in patients with bone metastases from solid tumours
Efficacy and safety of 120 mg XGEVA SC every 4 weeks or 4 mg zoledronic acid (dose-adjusted for reduced renal function) IV every 4 weeks were compared in three randomised, double blind, active controlled studies, in IV-bisphosphonate naïve patients with advanced malignancies involving bone: adults with breast cancer (study 1), other solid tumours or multiple myeloma (study 2), and castrate-resistant prostate cancer (study 3). Patients with prior history of ONJ or osteomyelitis of the jaw, an active dental or jaw condition requiring oral surgery, non-healed dental/oral surgery, or any planned invasive dental procedure, were not eligible for inclusion in these studies. The primary and secondary endpoints evaluated the occurrence of one or more skeletal related events (SREs). In studies demonstrating superiority of XGEVA to zoledronic acid, patients were offered open label XGEVA in a pre-specified 2-year extension treatment phase.
XGEVA reduced the risk of developing a SRE, and developing multiple SREs (first and subsequent) in patients with bone metastases from solid tumours (see table 2).
Table 2: Efficacy results in patients with advanced malignancies involving bone
|
Study 1
breast cancer |
Study 2
other solid tumours**
or multiple myeloma |
Study 3
prostate cancer |
Combined
advanced cancer |
| |
XGEVA |
zoledronic acid |
XGEVA |
zoledronic acid |
XGEVA |
zoledronic acid |
XGEVA |
zoledronic acid |
|
N |
1026 |
1020 |
886 |
890 |
950 |
951 |
2862 |
2861 |
|
First SRE |
|
Median time (months) |
NR |
26.4 |
20.6 |
16.3 |
20.7 |
17.1 |
27.6 |
19.4 |
|
Difference in median time (months) |
NA |
4.2 |
3.5 |
8.2 |
|
HR (95% CI) / RRR (%) |
0.82 (0.71, 0.95) / 18 |
0.84 (0.71, 0.98) / 16 |
0.82 (0.71, 0.95) / 18 |
0.83 (0.76, 0.90) / 17 |
|
Non-inferiority / Superiority p-values |
< 0.0001† / 0.0101† |
0.0007† / 0.0619† |
0.0002† / 0.0085† |
< 0.0001 / < 0.0001 |
|
Proportion of subjects (%) |
30.7 |
36.5 |
31.4 |
36.3 |
35.9 |
40.6 |
32.6 |
37.8 |
|
First and subsequent SRE* |
|
Mean number/patient |
0.46 |
0.60 |
0.44 |
0.49 |
0.52 |
0.61 |
0.48 |
0.57 |
|
Rate ratio (95% CI) / RRR (%) |
0.77 (0.66, 0.89) / 23 |
0.90 (0.77, 1.04) / 10 |
0.82 (0.71, 0.94) / 18 |
0.82 (0.75, 0.89) / 18 |
|
Superiority p-value |
0.0012† |
0.1447† |
0.0085† |
< 0.0001 |
|
SMR per Year |
0.45 |
0.58 |
0.86 |
1.04 |
0.79 |
0.83 |
0.69 |
0.81 |
|
First SRE or HCM |
|
Median time (months) |
NR |
25.2 |
19.0 |
14.4 |
20.3 |
17.1 |
26.6 |
19.4 |
|
HR (95% CI) / RRR (%) |
0.82 (0.70, 0.95) / 18 |
0.83 (0.71, 0.97) / 17 |
0.83 (0.72, 0.96) / 17 |
0.83 (0.76, 0.90) / 17 |
|
Superiority p-value |
0.0074 |
0.0215 |
0.0134 |
< 0.0001 |
|
First radiation to bone |
|
Median time (months) |
NR |
NR |
NR |
NR |
NR |
28.6 |
NR |
33.2 |
|
HR (95% CI) / RRR (%) |
0.74 (0.59, 0.94) / 26 |
0.78 (0.63, 0.97) / 22 |
0.78 (0.66, 0.94) / 22 |
0.77 (0.69, 0.87) / 23 |
|
Superiority p-value |
0.0121 |
0.0256 |
0.0071 |
< 0.0001 |
NR = not reached; NA = not available; HCM = hyper
calcaemia of malignancy; SMR = skeletal morbidity rate; HR = Hazard Ratio; RRR = Relative Risk Reduction †Adjusted p-values are presented for Studies 1, 2 and 3 (first SRE and first and subsequent SRE endpoints); *Accounts for all skeletal events over time; only events occurring ≥ 21 days after the previous event are counted.
** Including NSCLC, renal cell cancer, colorectal cancer, small cell lung cancer, bladder cancer, head and neck cancer, GI/genitourinary cancer and others, excluding breast and prostate cancer
Figure 1. Kaplan-Meier plots of time to first on-study SRE
ZA – Zoledronic Acid 4 mg Q4W
Dmab – Denosumab 120 mg Q4W
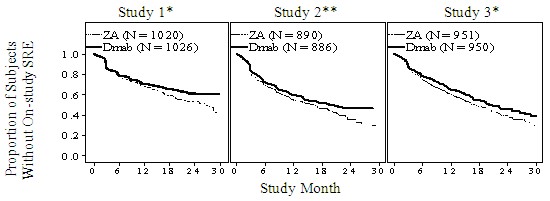
N = Number of subjects randomised
*= Statistically significant for superiority; **= Statistically significant for non-inferiority
Disease progression and overall survival
Disease progression was similar between XGEVA and zoledronic acid in all three studies and in the pre-specified analysis of all three-studies combined.
In all three studies overall survival was balanced between XGEVA and zoledronic acid in patients with advanced malignancies involving bone: patients with breast cancer (hazard ratio and 95% CI was 0.95 [0.81, 1.11]), patients with prostate cancer (hazard ratio and 95% CI was 1.03 [0.91, 1.17]), and patients with other solid tumours or multiple myeloma (hazard ratio and 95% CI was 0.95 [0.83, 1.08]). A post-hoc analysis in study 2 (patients with other solid tumours or multiple myeloma) examined overall survival for the 3 tumour types used for stratification (non-small cell lung cancer, multiple myeloma, and other). Overall survival was longer for XGEVA in non-small cell lung cancer (hazard ratio [95% CI] of 0.79 [0.65, 0.95]; n = 702) and longer for zoledronic acid in multiple myeloma (hazard ratio [95% CI] of 2.26 [1.13, 4.50]; n = 180) and similar between XGEVA and zoledronic acid in other tumour types (hazard ratio [95% CI] of 1.08 (0.90, 1.30); n = 894). This study did not control for prognostic factors and anti-neoplastic treatments. In a combined pre-specified analysis from studies 1, 2 and 3, overall survival was similar between XGEVA and zoledronic acid (hazard ratio and 95% CI 0.99 [0.91, 1.07]).
Effect on pain
The time to pain improvement (i.e., ≥ 2 point decrease from baseline in BPI-SF worst pain score) was similar for denosumab and zoledronic acid in each study and the integrated analyses. In a post-hoc analysis of the combined dataset, the median time to worsening pain (> 4-point worst pain score) in patients with mild or no pain at baseline was delayed for XGEVA compared to zoledronic acid (198 versus 143 days) (p = 0.0002).
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with XGEVA in all subsets of the paediatric population in prevention of skeletal related events in patients with bone metastases (see section 4.2 for information on paediatric use).
5.2 Pharmacokinetic properties
Absorption
Following SC administration, bioavailability was 62%.
Biotransformation
Denosumab is composed solely of amino acids and carbohydrates as native immunoglobulin and is unlikely to be eliminated via hepatic metabolic mechanisms. Its metabolism and elimination are expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides and individual amino acids.
Elimination
With multiple doses of 120 mg every 4 weeks an approximate 2-fold accumulation in serum denosumab concentrations was observed and steady-state was achieved by 6 months, consistent with time-independent pharmacokinetics. In subjects who discontinued 120 mg every 4 weeks, the mean half-life was 28 days (range 14 to 55 days).
A population pharmacokinetic analysis did not indicate clinically significant changes in the systemic exposure of denosumab at steady state with respect to age (18 to 87 years), race/ethnicity (Blacks, Hispanics, Asians and Caucasians explored), gender or solid tumour types. Increasing body weight was associated with decreases in systemic exposure, and vice versa. The alterations were not considered clinically relevant, since pharmacodynamic effects based on bone turnover markers were consistent across a wide range of body weight.
Linearity/non-linearity
Denosumab displayed non-linear pharmacokinetics with dose over a wide dose range, but approximately dose-proportional increases in exposure for doses of 60 mg (or 1 mg/kg) and higher. The non-linearity is likely due to a saturable target-mediated elimination pathway of importance at low concentrations.
Renal impairment
In a study of 55 patients without advanced cancer but with varying degrees of renal function, including patients on dialysis, the degree of renal impairment had no effect on the pharmacokinetics of denosumab. There is no need for renal monitoring when receiving XGEVA.
Hepatic impairment
No specific study in patients with hepatic impairment was performed. In general, monoclonal antibodies are not eliminated via hepatic metabolic mechanisms. The pharmacokinetics of denosumab is not expected to be affected by hepatic impairment.
Elderly
No overall differences in safety or efficacy were observed between geriatric patients and younger patients. Controlled clinical studies of XGEVA in patients with advanced malignancies involving bone over age 65 revealed similar efficacy and safety in older and younger patients. No dose adjustment is required in elderly patients.
Paediatric population
The pharmacokinetic profile in paediatric populations has not been assessed.
5.3 Preclinical safety data
Since the biological activity of denosumab in animals is specific to nonhuman primates, evaluation of genetically engineered (knockout) mice or use of other biological inhibitors of the RANK/RANKL pathway, such as OPG-Fc and RANK-Fc, were used to evaluate the pharmacodynamic properties of denosumab in rodent models.
In mouse bone metastasis models of oestrogen receptor positive and negative human breast cancer, prostate cancer and non small cell lung cancer, OPG-Fc reduced osteolytic, osteoblastic, and osteolytic/osteoblastic lesions, delayed formation of de novo bone metastases, and reduced skeletal tumour growth. When OPG-Fc was combined with hormonal therapy (tamoxifen) or chemotherapy (docetaxel) in these models, there was additive inhibition of skeletal tumour growth in breast, and prostate or lung cancer respectively. In a mouse model of mammary tumour induction, RANK-Fc reduced hormone-induced proliferation in mammary epithelium and delayed tumour formation.
Standard tests to investigate the genotoxicity potential of denosumab have not been evaluated, since such tests are not relevant for this molecule. However, due to its character it is unlikely that denosumab has any potential for genotoxicity.
The carcinogenic potential of denosumab has not been evaluated in long-term animal studies.
In single and repeated dose toxicity studies in cynomolgus monkeys, denosumab doses resulting in 2.7 to 15 times greater systemic exposure than the recommended human dose had no impact on cardiovascular physiology, male or female fertility, or produced specific target organ toxicity.
In a study of cynomolgus monkeys dosed with denosumab during the period equivalent to the first trimester of pregnancy, denosumab doses resulting in 9 times greater systemic exposure than the recommended human dose did not induce maternal toxicity or foetal harm during a period equivalent to the first trimester, although foetal lymph nodes were not examined.
In another study of cynomolgus monkeys dosed with denosumab throughout pregnancy at systemic exposures 12-fold higher than the human dose, there were increased stillbirths and postnatal mortality; abnormal bone growth resulting in reduced bone strength, reduced haematopoiesis, and tooth malalignment; absence of peripheral lymph nodes; and decreased neonatal growth. A no observed adverse effect level for reproductive effects was not established. Following a 6 month period after birth, bone related changes showed recovery and there was no effect on tooth eruption. However, the effects on lymph nodes and tooth malalignment persisted, and minimal to moderate mineralisation in multiple tissues was seen in one animal (relation to treatment uncertain).There was no evidence of maternal harm prior to labour; adverse maternal effects occurred infrequently during labour. Maternal mammary gland development was normal.
In preclinical bone quality studies in monkeys on long-term denosumab treatment, decreases in bone turnover were associated with improvement in bone strength and normal bone histology.
In male mice genetically engineered to express huRANKL (knock-in mice), which were subjected to a transcortical fracture, denosumab delayed the removal of cartilage and remodelling of the fracture callus compared to control, but biomechanical strength was not adversely affected.
In preclinical studies knockout mice lacking RANK or RANKL had an absence of lactation due to inhibition of mammary gland maturation (lobulo-alveolar gland development during pregnancy) and exhibited impairment of lymph node formation. Neonatal RANK/RANKL knockout mice exhibited decreased body weight, reduced bone growth, altered growth plates and lack of tooth eruption. Reduced bone growth, altered growth plates and impaired tooth eruption were also seen in studies of neonatal rats administered RANKL inhibitors, and these changes were partially reversible when dosing of RANKL inhibitor was discontinued. Adolescent primates dosed with denosumab at 2.7 and 15 times (10 and 50 mg/kg dose) the clinical exposure had abnormal growth plates. Therefore, treatment with denosumab may impair bone growth in children with open growth plates and may inhibit eruption of dentition.
6. Pharmaceutical particulars
6.1 List of excipients
Acetic acid, glacial*
Sodium hydroxide (for pH adjustment)*
Sorbitol (E420)
Water for injections
* Acetate buffer is formed by mixing acetic acid with sodium hydroxide
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.3 Shelf life
3 years.
XGEVA may be stored at room temperature (up to 25°C) for up to 30 days in the original container. Once removed from the refrigerator, XGEVA must be used within this 30 day period.
6.4 Special precautions for storage
Store in a refrigerator (2°C – 8°C).
Do not freeze.
Keep the vial in the outer carton in order to protect from light.
6.5 Nature and contents of container
1.7 ml solution in a single use vial (type I glass) with stopper (fluoropolymer coated elastomeric) and seal (aluminium) with flip-off cap.
Pack size of one, three or four.
Not all pack sizes may be marketed.
6.6 Special precautions for disposal and other handling
Before administration, the XGEVA solution should be inspected visually. The solution may contain trace amounts of translucent to white proteinaceous particles. Do not inject the solution if it is cloudy or discoloured. Do not shake excessively. To avoid discomfort at the site of injection, allow the vial to reach room temperature (up to 25°C) before injecting and inject slowly. Inject the entire contents of the vial. A 27 gauge needle is recommended for the administration of denosumab. Do not re-enter the vial.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
7. Marketing authorisation holder
Amgen Europe B.V.
Minervum 7061
NL-4817 ZK Breda
The Netherlands
8. Marketing authorisation number(s)
EU/1/11/703/001
EU/1/11/703/002
EU/1/11/703/003
9. Date of first authorisation/renewal of the authorisation
Date of first authorisation: 13 July 2011
10. Date of revision of the text
24 October 2013
Detailed information on this medicinal product is available on the website of the European Medicines Agency http://www.ema.europa.eu/.

FDA批准安进Xgeva骨巨细胞瘤新适应症
2013年6月13日,美国食品药品管理局(FDA)批准狄诺塞麦(Xgeva)新适应症,用于治疗成人及部分青少年患者的骨巨细胞瘤(GCTB),这是一种罕见而通常又是良性的肿瘤。
骨巨细胞瘤一般发生在20岁至40岁之间的成人患者身上。在大多数情况下,骨巨细胞瘤不会扩散到身体的其它部位,但会损害正常的骨生长,导致疼痛,活动范围受限和骨折。少数情况下,骨巨细胞瘤也能转变成一种恶性肿瘤,并扩散到肺部。
狄诺塞麦是一种与RANKL(一种保持骨骼健康的必需蛋白)相结合的单克隆抗体。RANKL也存在于骨巨细胞瘤中。狄诺塞麦用于骨巨细胞瘤不能通过手术切除(无法切除)或手术可能会导致严重病状(如截肢或关节切除)的患者。只有骨骼已经发育成熟的青少年患者才能使用这款药物。
FDA药品评价和研究中心血液学与肿瘤学产品部门主任,医学博士Richard Pazdur说:“今天狄诺塞麦新适应症的批准为不能进行手术或手术后会导致严重病状的骨巨细胞瘤患者提供了一种恰如所需的治疗选择。”FDA通过优先审评程序对这款药物进行了审评,而优先审评可以为药物提供一个加快的审评。狄诺塞麦因用于治疗罕见疾病或症状而被授予孤儿药产品资格。
狄诺塞麦用于骨巨细胞瘤的安全性和有效性基于两项总共由305名成人或青少年患者参与的临床试验。所有患者为确诊患有复发、不能切除或手术后会导致严重病状的骨巨细胞瘤病例。平均用药三个月之后,在187名肿瘤能够被测量的患者中,有47名患者的肿瘤出现萎缩。在平均超过20个月的随访后,有三位最初治疗期间肿瘤变小的患者,其骨巨细胞瘤又重新增长。
狄诺塞麦常见的副作用有关节痛、头痛、恶心、疲劳、背部及手足疼痛。最常见的严重副作用有颌骨坏死及骨髓炎(炎症或骨感染)。由于狄诺塞麦对胎儿有潜在伤害,育龄妇女使用这款药物时必须采取高度有效的避孕措施。
狄诺塞麦于2010被批准用于预防癌症已扩散至骨组织患者的骨折。这款药物由位于加利福尼亚州千橡市的安进公司上市销售。
-----------------------------------------------
产地国家: 美国
原产地英文商品名:
Xgeva 120mg/1.7mL(70mg/mL)/Vial
原产地英文药品名:
denosumab
中文参考商品译名:
Xgeva注射剂 120毫克/1.7毫升/瓶
中文参考药品译名:
狄诺塞麦
生产厂家中文参考译名:
安进
生产厂家英文名:
Amgen
|
