|
英文药名: Iressa(Gefitinib Tablets) 中文药名: 易瑞沙(吉非替尼片) 生产厂家: AstraZeneca
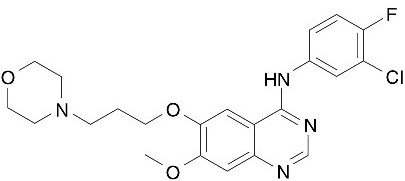
FOR ONCOLOGY USE ONLY DESCRIPTION IRESSA® (gefitinib tablets) contain 250 mg of gefitinib and are available as brown film-coated tablets for daily oral administration. Gefitinib is an anilinoquinazoline with the chemical name 4-Quinazolinamine, N-(3-chloro-4-fluorophenyl)-7-methoxy-6-[3-4-morpholin) propoxy] and the following structural formula:
It has the molecular formula C22H24ClFN4O3, a relative molecular mass of 446.9 and is a white-colored powder. Gefitinib is a free base. The molecule has pKas of 5.4 and 7.2 and therefore ionizes progressively in solution as the pH falls. Gefitinib can be defined as sparingly soluble at pH 1, but is practically insoluble above pH 7, with the solubility dropping sharply between pH 4 and pH 6. In non-aqueous solvents, gefitinib is freely soluble in glacial acetic acid and dimethylsulphoxide, soluble in pyridine, sparingly soluble in tetrahydrofuran, and slightly soluble in methanol, ethanol (99.5%), ethyl acetate, propan-2-ol and acetonitrile. The inactive ingredients of IRESSA tablets are: Tablet core: Lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, povidone, sodium lauryl sulfate and magnesium stearate. Coating: hypromellose, polyethylene glycol 300, titanium dioxide, red ferric oxide and yellow ferric oxide. CLINICAL PHARMACOLOGY Mechanism of Action The mechanism of the clinical antitumor action of gefitinib is not fully characterized. Gefitinib inhibits the intracellular phosphorylation of numerous tyrosine kinases associated with transmembrane cell surface receptors, including the tyrosine kinases associated with the epidermal growth factor receptor (EGFR-TK). EGFR is expressed on the cell surface of many normal cells and cancer cells. No clinical studies have been performed that demonstrate a correlation between EGFR receptor expression and response to gefitinib. Pharmacokinetics Gefitinib is absorbed slowly after oral administration with mean bioavailability of 60%. Elimination is by metabolism (primarily CYP3A4) and excretion in feces. The elimination half-life is about 48 hours. Daily oral administration of gefitinib to cancer patients resulted in a 2-fold accumulation compared to single dose administration. Steady state plasma concentrations are achieved within 10 days. Absorption and Distribution Gefitinib is slowly absorbed, with peak plasma levels occurring 3-7 hours after dosing and mean oral bioavailability of 60%. Bioavailability is not significantly altered by food. Gefitinib is extensively distributed throughout the body with a mean steady state volume of distribution of 1400 L following intravenous administration. In vitro binding of gefitinib to human plasma proteins (serum albumin and α1-acid glycoprotein) is 90% and is independent of drug concentrations. Metabolism and Elimination Gefitinib undergoes extensive hepatic metabolism in humans, predominantly by CYP3A4. Three sites of biotransformation have been identified: metabolism of the N-propoxymorpholino-group, demethylation of the methoxy-substituent on the quinazoline, and oxidative defluorination of the halogenated phenyl group. Five metabolites were identified in human plasma. Only O-desmethyl gefitinib has exposure comparable to gefitinib. Although this metabolite has similar EGFR-TK activity to gefitinib in the isolated enzyme assay, it had only 1/14 of the potency of gefitinib in one of the cell-based assays. Gefitinib is cleared primarily by the liver, with total plasma clearance and elimination half-life values of 595 mL/min and 48 hours, respectively, after intravenous administration. Excretion is predominantly via the feces (86%), with renal elimination of drug and metabolites accounting for less than 4% of the administered dose. Special Populations In population based data analyses, no relationships were identified between predicted steady state trough concentration and patient age, body weight, gender, ethnicity or creatinine clearance. Pediatric: Gefitinib has been evaluated in a Phase I pharmacokinetic trial in pediatric patients. In pediatric patients with refractory solid tumors the maximum tolerated dose (MTD) of gefitinib was determined to be 400 mg/m2 given orally once-daily. At this dose the median steady-state peak plasma concentration was 2.2 mg/ml and the median steady-state area under curve was 29.3 mg.h/ml. These values are comparable to those observed in adults at 700 mg/day. The most frequent toxicities observed were rash and altered ALT and AST. Hepatic Impairment: The influence of hepatic metastases with elevation of serum aspartate aminotransferase (AST/SGOT), alkaline phosphatase, and bilirubin has been evaluated in patients with normal (14 patients), moderately elevated (13 patients) and severely elevated (4 patients) levels of one or more of these biochemical parameters. Patients with moderately and severely elevated biochemical liver abnormalities had gefitinib pharmacokinetics similar to individuals without liver abnormalities (see PRECAUTIONS section). Renal Impairment: No clinical studies were conducted with IRESSA in patients with severely compromised renal function (see PRECAUTIONS section). Gefitinib and its metabolites are not significantly excreted via the kidney (<4%). Drug-Drug Interactions: In human liver microsome studies, gefitinib had no inhibitory effect on CYP1A2, CYP2C9, and CYP3A4 activities at concentrations ranging from 2-5000 ng/mL. At the highest concentration studied (5000 ng/mL), gefitinib inhibited CYP2C19 by 24% and CYP2D6 by 43%. Exposure to metoprolol, a substrate of CYP2D6, was increased by 30% when it was given in combination with gefitinib (500 mg daily for 28 days) in patients with solid tumors. The production of O-desmethyl gefitinib has been shown, in vitro, to be via CYP2D6. The role of CYP2D6 in the metabolic clearance of gefitinib has been evaluated in a clinical trial in healthy volunteers genotyped for CYP2D6 status. In poor metabolizers no measurable levels of O-desmethyl gefitinib were produced. The range of gefitinib exposures achieved in both the extensive and the poor metabolizer groups were wide and overlapping but the mean exposure to gefitinib was 2-fold higher in the poor metabolizer group. The higher average exposures that could be achieved by individuals with no active CYP2D6 may be clinically relevant since adverse experiences are related to dose and exposure. Rifampicin, an inducer of CYP3A4, reduced mean AUC of gefitinib by 85% in healthy male volunteers (see PRECAUTIONS-Drug Interactions and DOSAGE AND ADMINISTRATION-Dosage Adjustment sections). Concomitant administration of itraconazole (200 mg QD for 12 days), an inhibitor of CYP3A4, with gefitinib (250 mg single dose) to healthy male volunteers, increased mean gefitinib AUC by 88% (see PRECAUTIONS-Drug Interactionssection). Co-administration of high doses of ranitidine with sodium bicarbonate (to maintain the gastric pH above pH 5.0) reduced mean gefitinib AUC by 44% (see PRECAUTIONS-Drug Interactions section). International Normalized Ratio (INR) elevations and/or bleeding events have been reported in some patients taking warfarin while on IRESSA therapy. Patients taking warfarin should be monitored regularly for changes in prothrombin time or INR (see PRECAUTIONS-Drug Interactions and ADVERSE REACTIONS sections). Clinical Studies Non-Small Cell Lung Cancer (NSCLC): Refractory Disease Tumor Response Study - A multicenter clinical trial in the United States evaluated the tumor response rate of IRESSA 250 and 500 mg/day in patients with advanced non-small cell lung cancer whose disease had progressed after at least two prior chemotherapy regimens including a platinum drug and docetaxel. IRESSA was taken once daily at approximately the same time each day. Two hundred and sixteen patients received IRESSA, 102 (47%) and 114 (53%) receiving 250 mg and 500 mg daily doses, respectively. Study patient demographics and disease characteristics are summarized in Table 1. Forty-one percent of the patients had received two prior treatment regimens, 33% three prior treatment regimens, and 25% four or more prior treatment regimens. Effectiveness of IRESSA as third line therapy was determined in the 142 evaluable patients with documented disease progression on platinum and docetaxel therapies or who had had unacceptable toxicity on these agents.
Table 2 shows tumor response rates and response duration. The overall response rate for the 250 and 500 mg doses combined was 10.6% (95% CI: 6%, 16.8%). Response rates appeared to be highly variable in subgroups of the treated population: 5.1% (4/79) in males, 17.5% (11/63) in females, 4.6% (5/108) in previous or current smokers, 29.4% (10/34) in nonsmokers, 12.4% (12/97) with adenocarcinoma histology, and 6.7% (3/45) with other NSCLC histologies. Similar differences were seen in a multinational study in patients who had received 1 or 2 prior chemotherapy regimens, at least 1 of which was platinum-based. In responders, the median time from diagnosis to study randomization was 16.7 months (range 8 to 34 months).
Non-Small Cell Lung Cancer (NSCLC): Refractory Disease Survival Study - A double-blind, placebo-controlled parallel-group trial randomized 1692 patients with advanced NSCLC to receive either IRESSA 250 mg daily plus Best Supportive Care or placebo plus Best Supportive Care. Patients had received 1 or 2 prior chemotherapy regimens and had progressed while receiving or within 90 days of the last dose of chemotherapy or were intolerant to the most recent prior chemotherapy regimen. The two treatment arms were well-balanced for demographic and disease-related patient characteristics. The primary endpoint of the study was survival. IRESSA did not significantly prolong survival (stratified log rank HR 0.89, P=0.11, Median 5.6 vs 5.1 months for IRESSA and placebo, respectively). Non-Small Cell Lung Cancer (NSCLC); Studies of First-line Treatment in Combination with Chemotherapy - Two large trials were conducted in chemotherapy-naïve patients with stage III and IV non-small cell lung cancer. Two thousand one hundred thirty patients were randomized to receive IRESSA 250 mg daily, IRESSA 500 mg daily, or placebo in combination with platinum-based chemotherapy regimens. The chemotherapies given in these first-line trials were gemcitabine and cis-platinum (N=1093) or carboplatin and paclitaxel (N=1037). The addition of IRESSA did not demonstrate any increase, or trend toward such an increase, in tumor response rates, time to progression, or overall survival. INDICATIONS AND USAGE IRESSA is indicated as monotherapy for the continued treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of both platinum-based and docetaxel chemotherapies who are benefiting or have benefited from IRESSA. In light of positive survival data with other agents including another oral EGFR inhibitor, physicians should use other treatment options in advanced non-small cell lung cancer patient populations who have received one or two prior chemotherapy regimens and are refractory or intolerant to their most recent regimen. The effectiveness of IRESSA was initially based on objective response rates (see CLINICAL PHARMACOLOGY-Clinical Studies section). Subsequent studies intended to demonstrate an increase in survival have been unsuccessful. Specifically, results from a large placebo-controlled randomized trial in patients with advanced NSCLC who progressed while receiving or within 90 days of the last dose of chemotherapy or were intolerant to the most recent prior chemotherapy regimen, did not show an improvement in survival (see CLINICAL PHARMACOLOGY- Clinical Studies section). Results from two large, controlled, randomized trials in first-line treatment of non-small cell lung cancer showed no benefit from adding IRESSA to doublet, platinum-based chemotherapy. CONTRAINDICATIONS IRESSA is contraindicated in patients with severe hypersensitivity to gefitinib or to any other component of IRESSA. WARNINGS Pulmonary Toxicity Cases of interstitial lung disease (ILD) have been observed in patients receiving IRESSA at an overall incidence of about 1%. Approximately 1/3 of the cases have been fatal. Based on data from worldwide clinical studies, expanded access/compassionate use and post-marketing use the estimated reporting rate of ILD-type events overall is approximately 0.3% outside of Japan and approximately 3% in Japan. From a phase III double blind clinical trial (1692 patients) comparing IRESSA plus best supportive care (BSC) to placebo plus BSC in patients with advanced NSCLC who had received 1 or 2 prior chemotherapy regimens and were refractory or intolerant to their most recent regimen, the incidence of ILD-type events in the overall population was similar, and approximately 1% in both treatment arms. The majority of ILD-type events reported were from patients of Asian ethnicity and the ILD incidence among patients of Asian ethnicity receiving IRESSA therapy and placebo was similar, approximately 3% and 4% respectively. One ILD-type event was fatal, and this occurred in a patient receiving placebo. In a Post-Marketing Surveillance study in Japan (3350 patients) the reported rate of ILD-type events in patients receiving IRESSA was 5.8%. Reports have described the adverse event as interstitial pneumonia, pneumonitis and alveolitis. Patients often present with the acute onset of dyspnea, sometimes associated with cough or low-grade fever, often becoming severe within a short time and requiring hospitalization. ILD has occurred in patients who have received prior radiation therapy (31% of reported cases), prior chemotherapy (57% of reported patients), and no previous therapy (12% of reported cases). Patients with concurrent idiopathic pulmonary fibrosis whose condition worsens while receiving IRESSA have been observed to have an increased mortality compared to those without concurrent idiopathic pulmonary fibrosis. In the event of acute onset or worsening of pulmonary symptoms (dyspnea, cough, fever), IRESSA therapy should be interrupted and a prompt investigation of these symptoms should occur. If ILD is confirmed, IRESSA should be discontinued and the patient treated appropriately (see PRECAUTIONS-Information for Patients, ADVERSE REACTIONS and DOSAGE AND ADMINISTRATION-Dosage Adjustment sections). Pregnancy Category D IRESSA may cause fetal harm when administered to a pregnant woman. A single dose study in rats showed that gefitinib crosses the placenta after an oral dose of 5 mg/kg (30 mg/m2, about 1/5 the recommended human dose on a mg/m2 basis). When pregnant rats were treated with 5 mg/kg from the beginning of organogenesis to the end of weaning gave birth, there was a reduction in the number of offspring born alive. This effect was more severe at 20 mg/kg and was accompanied by high neonatal mortality soon after parturition. In this study a dose of 1 mg/kg caused no adverse effects. In rabbits, a dose of 20 mg/kg/day (240 mg/m2, about twice the recommended dose in humans on a mg/m2 basis) caused reduced fetal weight. There are no adequate and well-controlled studies in pregnant women using IRESSA. If IRESSA is used during pregnancy or if the patient becomes pregnant while receiving this drug, she should be apprised of the potential hazard to the fetus or potential risk for loss of the pregnancy. PRECAUTIONS Hepatotoxicity Asymptomatic increases in liver transaminases have been observed in IRESSA treated patients; therefore, periodic liver function (transaminases, bilirubin, and alkaline phosphatase) testing should be considered. Discontinuation of IRESSA should be considered if changes are severe. Patients with Hepatic Impairment In vitro and in vivo evidence suggest that gefitinib is cleared primarily by the liver. Therefore, gefitinib exposure may be increased in patients with hepatic dysfunction. In patients with liver metastases and moderately to severely elevated biochemical liver abnormalities, however, gefitinib pharmacokinetics were similar to the pharmacokinetics of individuals without liver abnormalities (see CLINICAL PHARMACOLOGY-Pharmacokinetics- Special Populations section). The influence of non-cancer related hepatic impairment on the pharmacokinetics of gefitinib has not been evaluated. Information for Patients Patients should be advised to seek medical advice promptly if they develop 1) severe or persistent diarrhea, nausea, anorexia, or vomiting, as these have sometimes been associated with dehydration; 2) an onset or worsening of pulmonary symptoms, ie, shortness of breath or cough; 3) an eye irritation; or, 4) any other new symptom (see WARNINGS-Pulmonary Toxicity, ADVERSE REACTIONS and DOSAGE AND ADMINISTRATION-Dosage Adjustment sections). Women of childbearing potential must be advised to avoid becoming pregnant (see WARNINGS-Pregnancy Category D). Drug Interactions Substances that are inducers of CYP3A4 activity increase the metabolism of gefitinib and decrease its plasma concentrations. In patients receiving a potent CYP3A4 inducer such as rifampicin or phenytoin, a dose increase to 500 mg daily should be considered in the absence of severe adverse drug reaction, and clinical response and adverse events should be carefully monitored (see CLINICAL PHARMACOLOGY-Pharmacokinetics-Drug-Drug Interactionsand DOSAGE AND ADMINISTRATION-Dosage Adjustment sections). International Normalized Ratio (INR) elevations and/or bleeding events have been reported in some patients taking warfarin while on IRESSA therapy. Patients taking warfarin should be monitored regularly for changes in prothrombin time or INR (see CLINICAL PHARMACOLOGY-Pharmacokinetics-Drug-Drug Interactions and ADVERSE REACTIONS sections). Substances that are potent inhibitors of CYP3A4 activity (eg, ketoconazole and itraconazole) decrease gefitinib metabolism and increase gefitinib plasma concentrations. This increase may be clinically relevant as adverse experiences are related to dose and exposure; therefore, caution should be used when administering CYP3A4 inhibitors with IRESSA (see CLINICAL PHARMACOLOGY-Pharmacokinetics-Drug-Drug Interactions and ADVERSE REACTIONS sections). Drugs that cause significant sustained elevation in gastric pH (histamine H2-receptor antagonists such as ranitidine or cimetidine) may reduce plasma concentrations of IRESSA and therefore potentially may reduce efficacy (see CLINICAL PHARMACOLOGY-Drug-Drug Interactions section). Phase II clinical trial data, where IRESSA and vinorelbine have been used concomitantly, indicate that IRESSA may exacerbate the neutropenic effect of vinorelbine. Carcinogenesis, Mutagenesis, Impairment of Fertility Gefitinib has been tested for genotoxicity in a series of in vitro (bacterial mutation, mouse lymphoma, and human lymphocyte) assays and an in vivo rat micronucleus test. Under the conditions of these assays, gefitinib did not cause genetic damage. A 2-year carcinogenicity study in rats resulted in a small but statistically significant increased incidence of hepatocellular adenomas in both male and female rats and mesenteric lymph node hemangiosarcomas in female rats at the highest dose (10 mg/kg/day) only. The hepatocellular adenomas were also seen in a 2- year carcinogenicity study in mice, which demonstrated a small increased incidence of this finding in male mice dosed at 50 mg/kg/day, and in both male and female mice at the highest dose of 90 mg/kg/day (reduced from 125 mg/kg/day from week 22). The effects reached statistical significance for the female mice, but not for the males. The clinical relevance of these findings is unknown. Pregnancy Pregnancy Category D(see WARNINGS and PRECAUTIONS-Information for Patients sections). Nursing Mothers It is not known whether IRESSA is excreted in human milk. Following oral administration of carbon-14 labeled gefitinib to rats 14 days postpartum, concentrations of radioactivity in milk were higher than in blood. Levels of gefitinib and its metabolites were 11-to-19-fold higher in milk than in blood, after oral exposure of lactating rats to a dose of 5 mg/kg. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants, women should be advised against breast-feeding while receiving IRESSA therapy. Pediatric Use IRESSA is not indicated for use in pediatric patients as safety and effectiveness have not been established (see CLINICAL PHARMACOLOGY-Special Populations-Pediatric). In clinical trials of IRESSA alone or with radiation in pediatric patients with primary Central Nervous System (CNS) tumors, cases of CNS hemorrhage and death have been reported. There are insufficient data in pediatric patients to establish a causal relationship. There is no evidence to suggest increased risk of cerebral hemorrhage in adult patients with NSCLC receiving IRESSA. Geriatric Use Of the total number of patients participating in trials of second- and third-line IRESSA treatment of NSCLC, 65% were aged 64 years or less, 30.5 % were aged 65 to 74 years, and 5% of patients were aged 75 years or older. No differences in safety or efficacy were observed between younger and older patients. Patients with Severe Renal Impairment The effect of severe renal impairment on the pharmacokinetics of gefitinib is not known. Patients with severe renal impairment should be treated with caution when given IRESSA. ADVERSE REACTIONS The safety database includes 941 patients from clinical trials and approximately 23,000 patients in the Expanded Access Program. Table 3 includes drug-related adverse events with an incidence of >5% for the 216 patients who received either 250 mg or 500 mg of IRESSA monotherapy for treatment of NSCLC. The most common adverse events reported at the recommended 250 mg daily dose were diarrhea, rash, acne, dry skin, nausea, and vomiting (see PRECAUTIONS-Information for Patients and DOSAGE AND ADMINISTRATION−Dosage Adjustment sections). The 500 mg dose showed a higher rate for most of these adverse events. Table 4 provides drug-related adverse events with an incidence of >5% by CTC grade for the patients who received the 250 mg/day dose of IRESSA monotherapy for treatment of NSCLC. In clinical trials, 2–3% of patients stopped therapy due to an adverse drug reaction (ADR). The onset of these ADRs occurred within the first month of therapy.
Other adverse events reported at an incidence of <5% in patients who received either 250 mg or 500 mg as monotherapy for treatment of NSCLC (along with their frequency at the 250 mg recommended dose) include the following: peripheral edema (2%), amblyopia (2%), dyspnea (2%), conjunctivitis (1%), vesiculobullous rash (1%), and mouth ulceration (1%). Interstitial Lung Disease Cases of interstitial lung disease (ILD) have been observed in patients receiving IRESSA at an overall incidence of about 1%. Approximately 1/3 of the cases have been fatal. Based on data from worldwide clinical studies, expanded access/compassionate use and post-marketing use the estimated reporting rate of ILD-type events overall is approximately 0.3% outside of Japan and approximately 3% in Japan. From a phase III double blind clinical trial (1692 patients) comparing IRESSA plus best supportive care (BSC) to placebo plus BSC in patients with advanced NSCLC who had received 1 or 2 prior chemotherapy regimens and were refractory or intolerant to their most recent regimen, the incidence of ILD-type events in the overall population was similar, and approximately 1% in both treatment arms. The majority of ILD-type events reported were from patients of Asian ethnicity and the ILD incidence among patients of Asian ethnicity receiving IRESSA therapy and placebo was similar, approximately 3% and 4% respectively. One ILD-type event was fatal, and this occurred in a patient receiving placebo. In a Post-Marketing Surveillance study in Japan (3350 patients) the reported rate of ILD-type events in patients receiving IRESSA was 5.8%. Reports have described the adverse event as interstitial pneumonia, pneumonitis and alveolitis. Patients often present with the acute onset of dyspnea, sometimes associated with cough or low-grade fever, often becoming severe within a short time and requiring hospitalization. ILD has occurred in patients who have received prior radiation therapy (31% of reported cases), prior chemotherapy (57% of reported patients), and no previous therapy (12% of reported cases). Patients with concurrent idiopathic pulmonary fibrosis whose condition worsens while receiving IRESSA have been observed to have an increased mortality compared to those without concurrent idiopathic pulmonary fibrosis. In the event of acute onset or worsening of pulmonary symptoms (dyspnea, cough, fever), IRESSA therapy should be interrupted and a prompt investigation of these symptoms should occur. If ILD is confirmed, IRESSA should be discontinued and the patient treated appropriately (see WARNINGS-Pulmonary Toxicity, PRECAUTIONS-Information for Patients and DOSAGE AND ADMINISTRATION-Dosage Adjustment sections). In patients receiving IRESSA therapy, there were reports of eye pain and corneal erosion/ulcer, sometimes in association with aberrant eyelash growth (see PRECAUTIONS-Information for Patients section). There have been reports of dry eyes, blepharitis and dry mouth, predominately mild in nature. Hemorrhage, such as epistaxis and hematuria have been reported in patients receiving IRESSA. There were also rare reports of pancreatitis and very rare reports of corneal membrane sloughing, ocular ischemia/hemorrhage, toxic epidermal necrolysis, Stevens-Johnson syndrome, erythema multiforme, and allergic reactions, including angioedema and urticaria. International Normalized Ratio (INR) elevations and/or bleeding events have been reported in some patients taking warfarin while on IRESSA therapy. Patients taking warfarin should be monitored regularly for changes in prothrombin time or INR (see CLINICAL PHARMACOLOGY-Drug-Drug Interactions and PRECAUTIONS-Drug Interactions sections). Asymptomatic elevations in blood creatinine have been reported. Data from non-clinical (in vitro and in vivo) studies indicate that gefitinib has the potential to inhibit the cardiac action potential repolarization process (eg, QT interval). The clinical relevance of these findings is unknown. OVERDOSAGE The acute toxicity of gefitinib up to 500 mg in clinical studies has been low. In non-clinical studies, a single dose of 12,000 mg/m2 (about 80 times the recommended clinical dose on a mg/m2 basis) was lethal to rats. Half this dose caused no mortality in mice. There is no specific treatment for an IRESSA overdose and possible symptoms of overdose are not established. However, in Phase 1 clinical trials, a limited number of patients were treated with daily doses of up to 1000 mg. An increase in frequency and severity of some adverse reactions was observed, mainly diarrhea and skin rash. Adverse reactions associated with overdose should be treated symptomatically; in particular, severe diarrhea should be managed appropriately. DOSAGE AND ADMINISTRATION The recommended daily dose of IRESSA is one 250 mg tablet with or without food. Higher doses do not give a better response and cause increased toxicity. For Patients who have Difficulty Swallowing Solids IRESSA tablets can also be dispersed in half a glass of drinking water (non-carbonated). No other liquids should be used. Drop the tablet in the water, without crushing it, stir until the tablet is dispersed (approximately 10 minutes) and drink the liquid immediately. Rinse the glass with half a glass of water and drink. The liquid can also be administered through a naso-gastric tube. Dosage Adjustment Patients with poorly tolerated diarrhea (sometimes associated with dehydration) or skin adverse drug reactions may be successfully managed by providing a brief (up to 14 days) therapy interruption followed by reinstatement of the 250 mg daily dose. In the event of acute onset or worsening of pulmonary symptoms (dyspnea, cough, fever), IRESSA therapy should be interrupted and a prompt investigation of these symptoms should occur and appropriate treatment initiated. If interstitial lung disease is confirmed, IRESSA should be discontinued and the patient treated appropriately (see WARNINGS-Pulmonary Toxicity, PRECAUTIONS-Information for Patients and ADVERSE REACTIONS sections). Patients who develop onset of new eye symptoms such as pain should be medically evaluated and managed appropriately, including IRESSA therapy interruption and removal of an aberrant eyelash if present. After symptoms and eye changes have resolved, the decision should be made concerning reinstatement of the 250 mg daily dose (see PRECAUTIONS-Information for Patients and ADVERSE REACTIONS sections). In patients receiving a potent CYP3A4 inducer such as rifampicin or phenytoin, a dose increase to 500 mg daily should be considered in the absence of severe adverse drug reaction, and clinical response and adverse events should be carefully monitored (see CLINICAL PHARMACOLOGY-Pharmacokinetics-Drug-Drug Interactions and PRECAUTIONS-Drug Interactions sections). No dosage adjustment is required on the basis of patient age, body weight, gender, ethnicity, or renal function; or in patients with moderate to severe hepatic impairment due to liver metastases (see CLINICAL PHARMACOLOGY-Pharmacokinetics-Special Populations section). HOW SUPPLIED IRESSA tablets are supplied as round, biconvex, brown film-coated tablets intagliated with "IRESSA 250" on one side and plain on the other side,each containing 250 mg of gefitinib. Bottles of 30 Tablets (NDC 0310-0482-30) Storage Store at controlled room temperature 20-25°C (68-77°F) [see USP]. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||