
|
近日,美国食品和药物管理局今天批准Daklinza(daclatasvir)用于与索非布韦治疗丙型肝炎病毒(HCV)基因型3感染。Daklinza是已经证明安全性和功效的治疗基因3的HCV感染,而不需要干扰素或利巴韦林共同给药的第一种药物,FDA批准的药物也被用于治疗HCV感染。
5 WARNINGS AND PRECAUTIONS
Lipase Elevations: Transient, asymptomatic lipase elevations of greater than 3 times the upper limit of normal (ULN) were observed in 2% of subjects in ALLY-3. 6.2 Postmarketing Experience Cardiac Disorders: Serious symptomatic bradycardia has been reported in patients taking amiodarone who initiate treatment with sofosbuvir in combination with another HCV direct-acting antiviral, including DAKLINZA [see Warnings and Precautions (5.2) and Drug Interactions (7.3)]. 7 DRUG INTERACTIONS 7.1 Potential for Other Drugs to Affect DAKLINZA Daclatasvir is a substrate of CYP3A. Therefore, moderate or strong inducers of CYP3A may decrease the plasma levels and therapeutic effect of daclatasvir [see Dosage and Administration (2.2), Contraindications (4), and Table 3]. Strong inhibitors of CYP3A (eg, clarithromycin, itraconazole, ketoconazole, ritonavir) may increase the plasma levels of daclatasvir [see Dosage and Administration (2.2) and Table 3]. 7.2 Potential for DAKLINZA to Affect Other Drugs Daclatasvir is an inhibitor of P-glycoprotein transporter (P-gp), organic anion transporting polypeptide (OATP) 1B1 and 1B3, and breast cancer resistance protein (BCRP). Administration of DAKLINZA may increase systemic exposure to medicinal products that are substrates of P-gp, OATP 1B1 or 1B3, or BCRP, which could increase or prolong their therapeutic effect or adverse reactions (see Table 3). 7.3 Established and Potentially Significant Drug Interactions Refer to the prescribing information for sofosbuvir for drug interaction information. The most conservative recommendation should be followed. Table 3 provides clinical recommendations for established or potentially significant drug interactions between DAKLINZA and other drugs [see Contraindications (4)]. Clinically relevant increase in concentration is indicated as “↑” and clinically relevant decrease as “↓” [for drug interaction data, see Clinical Pharmacology (12.3)]. Table 3: Established and Other Potentially Significant Drug Interactions
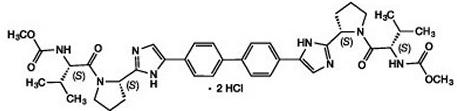
Based on the results of drug interaction trials [see Clinical Pharmacology (12.3)], no clinically relevant changes in exposure were observed for cyclosporine, escitalopram, ethinyl estradiol/norgestimate, methadone, midazolam, tacrolimus, or tenofovir with concomitant use of daclatasvir. No clinically relevant changes in daclatasvir exposure were observed with cyclosporine, escitalopram, famotidine, omeprazole, sofosbuvir, tacrolimus, or tenofovir. No clinically relevant interaction is anticipated for daclatasvir or the following concomitant medications: peginterferon alfa, ribavirin, or antacids. 8 USE IN SPECIFIC POPULATIONS 8.1 Pregnancy Risk Summary No data with DAKLINZA in pregnant women are available to inform a drug-associated risk. In animal reproduction studies in rats and rabbits, no evidence of fetal harm was observed with oral administration of daclatasvir during organogenesis at doses that produced exposures up to 6 and 22 times, respectively, the recommended human dose (RHD) of 60 mg. However, embryofetal toxicity was observed in rats and rabbits at maternally toxic doses that produced exposures of 33 and 98 times the human exposure, respectively, at the RHD of 60 mg [see Data]. Consider the benefits and risks of DAKLINZA when prescribing DAKLINZA to a pregnant woman. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. Data Animal Data Daclatasvir was administered orally to pregnant rats at doses of 0, 50, 200, or 1000 mg/kg/day on gestation days 6 to 15. Maternal toxicity (mortality, adverse clinical signs, body-weight losses, and reduced food consumption) was noted at doses of 200 and 1000 mg/kg/day. In the offspring, malformations of the fetal brain, skull, eyes, ears, nose, lip, palate, or limbs were observed at doses of 200 and 1000 mg/kg. The dose of 1000 mg/kg was associated with profound embryolethality and lower fetal body weight. No malformations were noted at 50 mg/kg/day. Systemic exposure (AUC) at 50 mg/kg/day in pregnant females was 6-fold higher than exposures at the RHD. In rabbits, daclatasvir was initially administered at doses of 0, 40, 200, or 750 mg/kg/day during the gestation days 7 to 19. Daclatasvir dosing was modified due to vehicle toxicity during the study to doses of 20, 99, and 370 mg/kg/day, respectively. Maternal toxicity was noted at doses of 200/99 and 750/370 mg/kg/day with adverse clinical signs and severe reductions in body weight and food consumption. Mortality and euthanasia occurred in multiple dams at 750/370 mg/kg/day. At 200/99 mg/kg/day, fetal effects included increased embryofetal lethality, reduced fetal body weights, and increased incidences of fetal malformations of the ribs as well as head and skull. No malformations were noted in rabbits at 40/20 mg/kg/day. Systemic exposures (AUC) at 40/20 mg/kg/day were 22-fold higher than exposures at the RHD. In a pre- and postnatal developmental study, daclatasvir was administered orally at 0, 25, 50, or 100 mg/kg/day from gestation day 6 to lactation day 20. At 100 mg/kg/day maternal toxicity included mortality and dystocia; developmental toxicity included slight reductions in offspring viability in the perinatal and neonatal periods and reductions in birth weight that persisted into adulthood. There was neither maternal nor developmental toxicity at doses up to 50 mg/kg/day. Systemic exposures (AUC) at this dose were 3.6-fold higher than the RHD. Daclatasvir was present in rat milk with concentrations 1.7- to 2-fold maternal plasma levels. 8.2 Lactation Risk Summary No information regarding the presence of daclatasvir in human milk, the effects on the breastfed infant, or the effects on milk production is available. Daclatasvir is present in the milk of lactating rats [see Use in Specific Populations (8.1)]. The development and health benefits of breastfeeding should be considered along with the mother’s clinical need for DAKLINZA and any potential adverse effects on the breastfed infant from DAKLINZA or from the underlying maternal condition. 8.4 Pediatric Use Safety and effectiveness of DAKLINZA in pediatric patients younger than 18 years of age have not been established. 8.5 Geriatric Use Safety was similar across older and younger subjects and there were no safety findings unique to subjects 65 years and older. Sustained virologic response (SVR) rates were comparable among older and younger subjects. No dosage adjustment of DAKLINZA is required for elderly patients [see Clinical Pharmacology (12.3)]. 8.6 Renal Impairment No dosage adjustment of DAKLINZA is required for patients with any degree of renal impairment [see Clinical Pharmacology (12.3)]. 8.7 Hepatic Impairment No dosage adjustment of DAKLINZA is required for patients with mild (Child-Pugh A), moderate (Child-Pugh B), or severe (Child-Pugh C) hepatic impairment [see Clinical Pharmacology (12.3)]. Safety and efficacy of DAKLINZA have not been established in patients with decompensated cirrhosis. 8.8 Liver Transplant Patients The safety and efficacy of DAKLINZA combination therapy have not been established in liver transplant patients. 10 OVERDOSAGE There is no known antidote for overdose of DAKLINZA. Treatment of overdose with DAKLINZA should consist of general supportive measures, including monitoring of vital signs and observation of the patient’s clinical status. Because daclatasvir is highly protein bound (>99%), dialysis is unlikely to significantly reduce plasma concentrations of the drug. 11 DESCRIPTION DAKLINZA (daclatasvir) is an inhibitor of HCV nonstructural protein 5A (NS5A). The chemical name for drug substance daclatasvir dihydrochloride is carbamic acid, N,N′-[[1,1′-biphenyl]-4,4′-diylbis[1H-imidazole-5,2-diyl-(2S)-2,1-pyrrolidinediyl[(1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl]]]bis-, C,C′-dimethyl ester, hydrochloride (1:2). Its molecular formula is C40H50N8O6•2HCl, and its molecular weight is 738.88 (free base). Daclatasvir dihydrochloride has the following structural formula:
In HCV-infected subjects following multiple oral doses of daclatasvir tablet ranging from 1 mg to 100 mg once daily, peak plasma concentrations occurred within 2 hours post dose. In vitro studies with human Caco-2 cells indicated that daclatasvir is a substrate of P-gp. The absolute bioavailability of the tablet formulation is 67%. Effect of Food on Oral Absorption In healthy subjects, administration of a daclatasvir 60 mg tablet after a high-fat, high-caloric meal (approximately 951 total kcal, 492 kcal from fat, 312 kcal from carbohydrates, 144 kcal from protein) decreased daclatasvir Cmax and AUC(0-inf) by 28% and 23%, respectively, compared with fasted conditions. A food effect was not observed with administration of a daclatasvir 60 mg tablet after a low-fat, low-caloric meal (approximately 277 total kcal, 41 kcal from fat, 190 kcal from carbohydrates, 44 kcal from protein) compared with fasted conditions [see Dosage and Administration (2)]. Distribution With multiple dosing, protein binding of daclatasvir in HCV-infected subjects was approximately 99% and independent of dose at the dose range studied (1-100 mg). In subjects who received daclatasvir 60 mg tablet orally followed by 100 μg [13C,15N]-daclatasvir intravenous dose, estimated volume of distribution at steady state was 47 L. Metabolism Daclatasvir is a substrate of CYP3A, with CYP3A4 being the primary CYP isoform responsible for metabolism. Following single-dose oral administration of 25 mg 14C-daclatasvir in healthy subjects, the majority of radioactivity in plasma was predominately attributed to parent drug (97% or greater). Elimination Following single-dose oral administration of 25 mg 14C-daclatasvir in healthy subjects, 88% of total radioactivity was recovered in feces (53% of the dose as unchanged daclatasvir) and 6.6% of the dose was excreted in the urine (primarily as unchanged daclatasvir). Following multiple-dose administration of daclatasvir in HCV-infected subjects, with doses ranging from 1 mg to 100 mg once daily, the terminal elimination half-life of daclatasvir ranged from approximately 12 to 15 hours. In subjects who received daclatasvir 60 mg tablet orally followed by 100 μg [13C,15N]-daclatasvir intravenous dose, the total clearance was 4.2 L/h. Specific Populations Renal Impairment The pharmacokinetics of daclatasvir following a single 60 mg oral dose was studied in non–HCV-infected subjects with renal impairment. Using a regression analysis, the predicted AUC(0‑inf) of daclatasvir was estimated to be 26%, 60%, and 80% higher in subjects with creatinine clearance (CLcr) values of 60, 30, and 15 mL/min, respectively, relative to subjects with normal renal function (CLcr of 90 mL/min, defined using the Cockcroft-Gault CLcr formula), and daclatasvir unbound AUC(0-inf) was predicted to be 18%, 39%, and 51% higher for subjects with CLcr values of 60, 30, and 15 mL/min, respectively, relative to subjects with normal renal function. Using observed data, subjects with end-stage renal disease requiring hemodialysis had a 27% increase in daclatasvir AUC(0-inf) and a 20% increase in unbound AUC(0-inf) compared to subjects with normal renal function as defined using the Cockcroft-Gault CLcr formula. [See Use in Specific Populations (8.6).] Daclatasvir is highly protein bound to plasma proteins and is unlikely to be removed by dialysis. Hepatic Impairment The pharmacokinetics of daclatasvir following a single 30 mg oral dose was studied in non–HCV-infected subjects with mild (Child-Pugh A), moderate (Child-Pugh B), and severe (Child-Pugh C) hepatic impairment compared to a corresponding matched control group. The Cmax and AUC(0-inf) of total daclatasvir (free and protein-bound drug) were lower by 46% and 43%, respectively, in Child-Pugh A subjects; by 45% and 38%, respectively, in Child-Pugh B subjects; and by 55% and 36%, respectively, in Child-Pugh C subjects. The Cmax and AUC(0‑inf) of unbound daclatasvir were lower by 43% and 40%, respectively, in Child-Pugh A subjects; by 14% and 2%, respectively, in Child-Pugh B subjects; and by 33% and 5%, respectively, in Child-Pugh C subjects [see Use in Specific Populations (8.7)]. Geriatric Population pharmacokinetic analysis in HCV-infected subjects showed that within the age range (18-79 years) analyzed, age did not have a clinically relevant effect on the pharmacokinetics of daclatasvir [see Use in Specific Populations (8.5)]. Pediatric and Adolescent The pharmacokinetics of daclatasvir in pediatric patients has not been evaluated. Gender Population pharmacokinetic analyses in HCV-infected subjects estimated that female subjects have a 30% higher daclatasvir AUC compared to male subjects. This difference in daclatasvir AUC is not considered clinically relevant. Race Population pharmacokinetic analyses in HCV-infected subjects indicated that race had no clinically relevant effect on daclatasvir exposure. Drug Interactions Cytochrome P450 (CYP) Enzymes Daclatasvir is a substrate of CYP3A. In vitro, daclatasvir did not inhibit (IC50 >40 µM) CYP enzymes 1A2, 2B6, 2C8, 2C9, 2C19, or 2D6. Daclatasvir did not have a clinically relevant effect on the exposure of midazolam, a sensitive CYP3A substrate. Transporters Daclatasvir is a substrate of P-gp. However, cyclosporine, which inhibits multiple transporters including P-gp, did not have a clinically relevant effect on the pharmacokinetics of daclatasvir. Daclatasvir, in vitro, did not inhibit organic cation transporter (OCT) 2 and did not have a clinically relevant effect on the pharmacokinetics of tenofovir, an organic anion transporter (OAT) substrate. Daclatasvir demonstrated inhibitory effects on digoxin (a P-gp substrate) and rosuvastatin (an OATP 1B1, OATP 1B3, and BCRP substrate) in drug-drug interaction trials. Drug interaction studies were conducted with daclatasvir and other drugs likely to be coadministered or drugs used as probes to evaluate potential drug-drug interactions. The effects of daclatasvir on the Cmax, AUC, and Cmin of the coadministered drug are summarized in Table 5, and the effects of the coadministered drug on the Cmax, AUC, and Cmin of daclatasvir are summarized in Table 6. For information regarding clinical recommendations, see Contraindications (4) and Drug Interactions (7.3). Drug interaction studies were conducted in healthy adults unless otherwise noted. Table 5: Effect of DAKLINZA on the Pharmacokinetics of Concomitant Drugs
Mechanism of Action Daclatasvir is an inhibitor of NS5A, a nonstructural protein encoded by HCV. Daclatasvir binds to the N-terminus of NS5A and inhibits both viral RNA replication and virion assembly. Characterization of daclatasvir-resistant viruses, biochemical studies, and computer modeling data indicate that daclatasvir interacts with the N-terminus within Domain 1 of the protein, which may cause structural distortions that interfere with NS5A functions. Antiviral Activity Daclatasvir had a median EC50 value of 0.2 nM (range, 0.006-3.2 nM, n=17) against hybrid replicons containing genotype 3a subject-derived NS5A sequences without detectable daclatasvir resistance-associated polymorphisms at NS5A amino acid positions 28, 30, 31, or 93. Daclatasvir activity was reduced against genotype 3a subject-derived replicons with resistance-associated polymorphisms at positions 28, 30, 31, or 93, with a median EC50 value of 13.5 nM (range, 1.3-50 nM). Similarly, the EC50 values of daclatasvir against 3 genotype 3b and 1 genotype 3i subject-derived NS5A sequences with polymorphisms (relative to a genotype 3a reference) at positions 30 or 31 were ≥3620 nM. The median EC50 values of daclatasvir for genotypes 1a, 1b, 2, 4, and 5 subject-derived NS5A hybrid replicons were 0.008 nM (range, 0.002-2409 nM, n=40), 0.002 nM (range, 0.0007-10 nM, n=42), 16 nM (range, 0.005-60 nM, n=16), 0.025 nM (range, 0.001-158 nM, n=14), and 0.004 nM (range, 0.003-0.019 nM, n=3), respectively. The EC50 value against a single HCV genotype 6 derived replicon was 0.054 nM. Daclatasvir was not antagonistic with interferon alfa, HCV NS3/4A protease inhibitors, HCV NS5B nucleoside analog inhibitors, and HCV NS5B non-nucleoside inhibitors in cell culture combination antiviral activity studies using the cell-based HCV replicon system. Resistance In Cell Culture HCV genotype 3a replicon variants with reduced susceptibility to daclatasvir were selected in cell culture, and the genotype and phenotype of daclatasvir-resistant variants were characterized. Phenotypic analysis of stable replicon cell lines showed that variant replicons containing A30K, A30T, L31F, S62L, and Y93H substitutions exhibited 56-, 1-, 603-, 1.75-, and 2737-fold reduced susceptibility to daclatasvir, respectively. In Clinical Studies Of 152 HCV genotype 3-infected subjects treated in the ALLY-3 trial, 17 experienced virologic failure, of whom 12 had cirrhosis. Post-baseline NS5A and NS5B population nucleotide sequencing data were available for virus from 17/17 and 16/17 subjects, respectively. Virus from all 17 subjects at the time of virologic failure harbored one or more of the NS5A resistance-associated substitutions A30K/S, L31I, S62A/L/P/T, or Y93H. The most common substitution at failure was Y93H (15/17 subjects), which was observed at baseline in 6 subjects and emerged in 9 subjects. For NS5B, 1 of 16 subjects had virus with the emergent NS5B resistance-associated substitution S282T at failure. Persistence of Resistance-Associated Substitutions Limited data from ALLY-3 on the persistence of daclatasvir resistance-associated substitutions in HCV genotype 3-infected subjects are available. In a separate long-term follow-up study of predominately HCV genotype 1-infected subjects treated with daclatasvir-containing regimens in phase 2/3 clinical trials, viral populations with treatment-emergent NS5A resistance-associated substitutions persisted at detectable levels for more than 1 year in most subjects. Effect of Baseline HCV Polymorphisms on Treatment Response In an analysis of 148 subjects with available baseline resistance data in ALLY-3, virus from 52% (77/148) of subjects had baseline NS5A polymorphisms at resistance-associated positions (defined as any change from reference at NS5A amino acid positions 28, 30, 31, 58, 62, 92, or 93) identified by population sequencing. The Y93H polymorphism was detected in 9% (13/148) of subjects receiving DAKLINZA and sofosbuvir and was associated with reduced SVR12 rates (Table 7). Polymorphisms detected at other NS5A resistance-associated positions were not associated with reduced SVR12 rates; these polymorphisms included M28V (n=1), A30K/S/T/V (n=14), P58R/S (n=3), and S62-any (n=66). Polymorphisms at positions associated with sofosbuvir resistance or exposure (defined as any change from reference at NS5B positions L159, S282, C316, L320, or V321) were not detected in the baseline NS5B sequence of any subject (n=150) in ALLY-3 by population-based sequencing. Phylogenetic analysis of NS5A sequences indicated that all subjects with available data (n=148) were infected with HCV subtype 3a. Table 7: SVR12 Rates in Subjects with HCV Genotype 3 with/without the Baseline NS5A Y93H Polymorphism, by Cirrhosis Status
Based on resistance patterns observed in cell culture replicon studies and HCV genotype 3-infected subjects, cross-resistance between daclatasvir and other NS5A inhibitors is expected. Cross-resistance between daclatasvir and other classes of direct-acting antivirals is not expected. The impact of prior daclatasvir treatment experience on the efficacy of other NS5A inhibitors has not been studied. Conversely, the efficacy of DAKLINZA in combination with sofosbuvir has not been studied in subjects who have previously failed treatment with regimens that include an NS5A inhibitor. 13 NONCLINICAL TOXICOLOGY 13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility Carcinogenesis and Mutagenesis A 2-year carcinogenicity study in Sprague Dawley rats and a 6-month study in transgenic (Tg rasH2) mice were conducted with daclatasvir. In the 2-year study in rats, no drug-related increase in tumor incidence was observed at doses up to 50 mg/kg/day (both sexes). Daclatasvir exposures at these doses were approximately 6-fold (males and females) the human systemic exposure at the therapeutic daily dose. In transgenic mice no drug-related increase in tumor incidence was observed at doses of 300 mg/kg/day (both sexes). Daclatasvir was not genotoxic in a battery of in vitro or in vivo assays, including bacterial mutagenicity (Ames) assays, mammalian mutation assays in Chinese hamster ovary cells, or in an in vivo oral micronucleus study in rats. Impairment of Fertility Daclatasvir had no effects on fertility in female rats at any dose tested. Daclatasvir exposures at these doses in females were approximately 24-fold the human systemic exposure at the therapeutic daily dose. In male rats, effects on reproductive endpoints at 200 mg/kg/day included reduced prostate/seminal vesicle weights, minimally increased dysmorphic sperm, as well as increased mean pre-implantation loss in litters sired by treated males. Daclatasvir exposures at the 200 mg/kg/day dose in males were approximately 26-fold the human systemic exposure at the therapeutic daily dose. Exposures at 50 mg/kg/day in males produced no notable effects and was 4.7-fold the exposure in humans at the recommended daily dose. 14 CLINICAL STUDIES The efficacy and safety of DAKLINZA in combination with sofosbuvir were evaluated in the phase 3 ALLY-3 (AI444-218) clinical trial. ALLY-3 was an open-label trial that included 152 subjects with chronic HCV genotype 3 infection and compensated liver disease who were treatment-naive (n=101) or treatment-experienced (n=51). Most treatment-experienced subjects had failed prior treatment with peginterferon/ribavirin, but 7 subjects had been treated previously with a sofosbuvir regimen and 2 subjects with a regimen containing an investigational cyclophilin inhibitor. Previous exposure to NS5A inhibitors was prohibited. Subjects received DAKLINZA 60 mg plus sofosbuvir 400 mg once daily for 12 weeks and were monitored for 24 weeks post treatment. HCV RNA values were measured during the clinical trial using the COBAS® TaqMan® HCV test (version 2.0), for use with the High Pure System. The assay had a lower limit of quantification (LLOQ) of 25 IU per mL. Sustained virologic response (SVR) was the primary endpoint and was defined as HCV RNA below the LLOQ at post-treatment week 12 (SVR12). The 152 treated subjects in ALLY-3 had a median age of 55 years (range, 24-73); 59% of the subjects were male; 90% were white, 5% were Asian, and 4% were black. Most subjects (76%) had baseline HCV RNA levels greater than or equal to 800,000 IU/mL; 21% of the subjects had compensated cirrhosis, and 40% had the IL28B rs12979860 CC genotype. SVR and outcomes in subjects without SVR in ALLY-3 are shown by patient population in Table 8. For SVR outcomes related to the baseline NS5A Y93H polymorphism, see Microbiology (12.4). SVR rates were comparable regardless of age, gender, IL28B allele status, or baseline HCV RNA level. Table 8: Treatment Outcomes in ALLY-3: DAKLINZA in Combination with Sofosbuvir in Subjects with HCV Genotype 3 Infection
16.1 How Supplied DAKLINZA is packaged in bottles as described in the table.
16.2 Storage Store DAKLINZA tablets at 25°C (77°F), with excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature]. 17 PATIENT COUNSELING INFORMATION Advise the patient to read the FDA-approved patient labeling (Patient Information). Drug Interactions Inform patients of the potential for drug interactions with DAKLINZA, and that some drugs should not be taken with DAKLINZA [see Contraindications (4), Drug Interactions (7), and Clinical Pharmacology (12.3)]. Symptomatic Bradycardia When Used in Combination with Sofosbuvir and Amiodarone Advise patients to seek medical evaluation immediately for symptoms of bradycardia, such as near-fainting or fainting, dizziness or lightheadedness, malaise, weakness, excessive tiredness, shortness of breath, chest pain, confusion or memory problems [see Warnings and Precautions (5.2), Adverse Reactions (6.2), and Drug Interactions (7.3)]. DAKLINZA Combination Therapy with Sofosbuvir Inform patients that DAKLINZA should not be used alone to treat genotype 3 chronic hepatitis C infection. DAKLINZA should be used in combination with sofosbuvir for the treatment of genotype 3 HCV infection [see Indications and Usage (1)]. Missed Doses Instruct patients that if they miss a dose of DAKLINZA, the dose should be taken as soon as possible if remembered within the same day. However, if the missed dose is not remembered within the same day, the dose should be skipped and the next dose taken at the appropriate time. For instructions for missed doses of other agents in the regimen, refer to the respective prescribing information. Hepatitis C Virus Transmission Inform patients that the effect of treatment of hepatitis C infection on transmission is not known, and that appropriate precautions to prevent transmission of the hepatitis C virus during treatment should be taken. BMS丙肝药物Daklinza的复方药物获欧盟批准 2014年8月27日,百时美施贵宝表示,欧洲盟委员会批准Daklinza (daclatasvir)与其它药物组成的复方药物用于整个基因型1、2、3和4慢性丙型肝炎病毒感染成人患者治疗。该公司指出,这款口服药物是在欧洲批准的首款NS5A复合抑制剂,可供与其它治疗药物组成复方药物使用,持续治疗周期为12周或24周,而以干扰素及利巴韦林为基础的方案治疗周期为48周。 6月份时,欧洲药品管理局人用医药产品委员会对这款药物给出了积极的意见,这次的批准由大量临床研究支持,包括一项Daklinza与吉利德科学Sovaldi (sofosbuvir)组成复方用于基因型1、2和3的研究。 百时美施贵宝指出,结果显示在治疗结束12周后,在99%未经治疗的HCV基因型1患者、100%的以Vertex制药特拉匹韦或默沙东波普瑞韦治疗失败的基因型1患者、96%的基因型2患者及89%的基因型3患者中,Daklinza与Sovaldi复方药物达到了持续病毒学响应。 百时美施贵宝全球商业主管Blin评论称,“我们期待继续与欧盟卫生监管机构一起确保以Daklinza为基础的方案尽可能快地用于患者。”Daklinza于7月份时获批与NS3/4A蛋白酶抑制剂Sunvepra (asunaprevir)组成的复方药物为该国首个全口服、无干扰素及利巴韦林治疗方案,用于慢性HCV感染基因型1患者,包括患有补偿性肝硬化患者。 2月份,FDA授予百时美施贵宝Daklinza与Asunaprevir的复方药物突破性治疗药物资格,该复方药物于4月份被提交上市申请,目标审评日期为11月30日。 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Daklinza Tablets(Daclatasvir Hydrochloride)简介:
近日,美国食品和药物管理局今天批准Daklinza(daclatasvir)用于与索非布韦治疗丙型肝炎病毒(HCV)基因型3感染。Daklinza是已经证明安全性和功效的治疗基因3的HCV感染,而不需要干扰素或利巴韦林共同给 ... 责任编辑:admin |
最新文章更多推荐文章更多热点文章更多
|

中国新特药网-第一时间提供全球医药资讯
注: 本站药品信息仅供医药研发机构作参考-
在线咨询:
- 鲁ICP备05035203号 @