|
麦罗塔Mylotarg(gemtuzumab)是奇霉素吉妥组单抗的合成制剂,是以抗体为基质的抗癌药物。该药是首个利用单克隆抗体技术研制的直接靶向癌细胞的化疗药物,目前主要用于治疗急性骨髓性白血病AML。
【商 品 名】Mylotarg
【通 用 名】gemtuzumab 吉妥单抗
【中 文 名】麦罗塔
【开发公司】惠氏、Wyeth CellTech
【销售公司】惠氏、Wyeth CellTech
【药物种类】Umab人流抗体;化疗药物卡奇霉素以gemtuzumab为载体。
【靶 点】CD—33抗体。
【适 应 症】急性髓性白血病,骨髓增生异常综合症。
【临床阶段】美国FDA2002年上市。
【作用机制】CD33 单抗与卡奇霉素(Calicheamicin)偶合,通过单抗将与CD33结合内化进入肿瘤细胞,在溶酶体中将药物释放出发挥作用。(注,肿瘤细胞产生耐药后,此药无效)
【贮藏条件】避光2℃—8℃,避免剧烈摇晃。
【规 格】 针剂。
【剂 型】5mg×1支/盒。
【生产商】美国惠氏
Polvo liofilizado para reconstituir a solución inyectable
Agente quimioterapéutico
(Gemtuzumab ozogamicina)
COMPOSICION Y CARACTERISTICAS FARMACEUTICAS: 1 vial contiene, como ingrediente activo, 5 mg de gemtuzumab ozogamicina.
Ingredientes no activos: Dextran 40; sacarosa cloruro de sodio; fosfato de sodio monobásico y dibásico.
DESCRIPCION
Principio activo: Gemtuzumab ozogamicina.
El gemtuzumab ozogamicina está compuesto de una IgG4 recombinante humanizada, anticuerpo kapa conjugado con un antibiótico citotóxico antitumoral, caliqueamicina, aislado de la fermentación de una bacteria, Micromonospora echinospora subsp. Calichensis. La porción anticuerpo de MYLOTARG® se une específicamente al antígeno CD33, una proteína de adhesión siálico ácido-dependiente encontrada en la superficie de los blastos leucémicos y en las células normales inmaduras de la línea mielomonocítico, pero no sobre las células madre hematopoyéticas normales. El anticuerpo anti-CD33 hP67.6 es producido del cultivo en suspensión de una célula de mamífero usando una línea de células de mieloma NS0 y purificado bajo condiciones que remueven o inactivan los virus. Tres pasos separados e independientes en el proceso de purificación del anticuerpo hP67.6 llevan a la inactivación y remoción del retrovirus. Estos pasos incluyen tratamiento de pH bajo, cromatografía DEAE-Sepharose y filtración viral. MYLOTARG® contiene secuencias de aminoácidos de los cuales aproximadamente el 98.3% son de origen humano. La región constante y las regiones marco contienen secuencias humanas, mientras que las regiones que determinan las complementarias se derivan de un anticuerpo de murina (p67.6) que enlaza el CD33. Este anticuerpo está ligado a la caliqueamicina N-acetil-gama por medio de un enlace bifuncional. El gemtuzumab ozogamicin tiene aproximadamente 50% del anticuerpo cargado con 4-6 moles de caliqueamicina por mol del anticuerpo. El restante 50% no está ligado al derivado caliqueamicina.
Peso molecular: Gemtuzumab ozogamicina tiene un peso molecular de 151 a 153 kDa.
Características físicas: MYLOTARG® es un polvo estéril, blanco, liofilizado, libre de preservativos. El producto es sensible a la luz y debe protegerse de la luz directa e indirecta del sol y de la luz fluorescente sin filtro durante la preparación y administración de la infusión.
Clase farmacológica, clase terapéutica: Agente quimioterapéutico.
Forma farmacéutica y vía de administración: Polvo liofilizado para solución por infusión.
Naturaleza y contenido del envase: Viales vidrio ámbar tipo I, tapón de goma y sello rompible.
INDICACIONES: MYLOTARG® está indicado para el tratamiento de los pacientes con leucemia mieloide aguda positiva para CD33 en su primera recaída de personas de 60 años o mayores y quienes no se consideran candidatos para otra quimioterapia citotóxica. No se ha establecido la seguridad y eficacia de MYLOTARG® en pacientes con desempeño pobre y desorden orgánico.
La efectividad de MYLOTARG® está basada en las tasas de respuesta total (TRT) (ver Farmacodinamia, eficacia clínica). No existen estudios clínicos controlados que demuestren un beneficio clínico, tal como mejoramiento de los síntomas relacionados con la enfermedad o aumento de la supervivencia, comparado con cualquier otro tratamiento.
DOSIS Y ADMINISTRACION
Dosis: La dosis recomendada de MYLOTARG® es 9 mg/m2 en infusión al menos durante un periodo de 2 horas (ver tambiénAlmacenamiento para información sobre dilución, almacenamiento e infusión). Los médicos deben considerar la leucorreducción con hidroxiurea o leucoferesis para reducir el recuento de los glóbulos blancos periféricos por debajo de 30.000/µL antes de la administración de MYLOTARG®. Se deben tomar las medidas apropiadas (ej.: hidratación y alopurinol) para prevenir hiperuricemia.
Los pacientes deben recibir los siguientes medicamentos profilácticos una hora antes de la administración de MYLOTARG®: difenhidramina vía oral y acetaminofén vía oral; luego, dos dosis adicionales de acetaminofén vía oral, una cada cuatro horas a medida que se necesite. Los signos vitales deben ser supervisados durante la infusión y durante las cuatro horas siguientes a la misma. El curso recomendado del tratamiento con MYLOTARG® es un total de 2 dosis con 14 días de intervalo entre dosis. No se requiere una recuperación completa de la toxicidad hematológica para la administración de la segunda dosis.
Los pacientes con deterioro renal o hepático no fueron incluidos en los estudios clínicos (ver Advertencias especiales y Precauciones).
Administración: MYLOTARG® NO DEBE SER ADMINISTRADO COMO UNA DOSIS DE CARGA O DOSIS UNICA INTRAVENOSA (IV).
La solución de MYLOTARG® reconstituida y diluida debe ser infundida al menos durante un periodo de 2 horas (ver también Almacenamiento para información sobre dilución, almacenamiento e infusión). MYLOTARG® puede ser administrado periféricamente o por medio de una línea central. Durante la infusión sólo la bolsa de la mezcla necesita ser protegida de la luz. Para la infusión debe utilizarse una línea equipada con un filtro terminal que enlaza proteínas. Las siguientes membranas de filtro son calificadas para este propósito: sulfona poliéter 0,22 micras o 1,2 micras (PES) (Supor®); filtro hidrofílico acrílico copolímero 1,2 micras (Versapor®); membrana de celulosa y éster (acetato o nitrato) 0,8 micras; membrana acetato celulosa 0,2 micras. NO COADMINISTRAR OTROS MEDICAMENTOS POR LA MISMA LINEA DE INFUSION.
Para instrucciones de manejo, reconstitución, dilución y protección contra la luz durante la infusión, ver la sección Manipulación.
CONTRAINDICACIONES: MYLOTARG® está contraindicado en pacientes con hipersensibilidad conocida a:
- Gemtuzumab ozogamicin o a cualquiera de sus componentes (anticuerpo anti-CD33 [hP67.6], o los derivados de la caliqueamicina
- Cualquiera de sus ingredientes no activos
ADVERTENCIAS ESPECIALES
Advertencias: MYLOTARG® debe ser administrado bajo la supervisión de médicos con experiencia en el tratamiento de la leucemia aguda y en instalaciones equipadas para supervisar y tratar a los pacientes con leucemia.
No existen estudios clínicos controlados que demuestren la eficacia y seguridad del uso de MYLOTARG® en combinación con otros agentes quimioterapéuticos. Por lo tanto, MYLOTARG® debe ser usado únicamente como quimioterapia de un agente único y no en regímenes de quimioterapia de combinación fuera de los estudios clínicos.
Se presenta mielosupresión severa cuando se usa MYLOTARG® en las dosis recomendadas.
Reacciones de hipersensibilidad incluyendo anafilaxis, reacciones a la infusión, eventos pulmonares: La administración de MYLOTARG® puede tener como resultado reacciones severas de hipersensibilidad (incluyendo anafilaxis), y otras reacciones relacionadas con la infusión que pueden incluir severos eventos pulmonares. Rara vez han sido fatales las reacciones de hipersensibilidad y los eventos pulmonares. En la mayoría de los casos, los síntomas relacionados con la infusión durante ésta o durante las 24 horas de la administración de MYLOTARG® ocurrieron y se resolvieron. La infusión de MYLOTARG® podría ser interrumpida en los pacientes que experimentan disnea o hipotensión clínicamente significativa. Se debe supervisar a los pacientes hasta que los signos y síntomas se hayan resuelto completamente. La descontinuación del tratamiento con MYLOTARG® podría ser considerada en pacientes que desarrollan anafilaxis, edema pulmonar o síndrome de dificultad respiratoria aguda. Ya que los pacientes con recuentos altos de blastos periféricos pueden tener un riesgo más grande de eventos pulmonares y para el síndrome de lisis tumoral, los médicos deben considerar la leucorreducción con hidroxiurea o leucoferesis para reducir el recuento de los glóbulos blancos periféricos por debajo de 30.000/µL antes de la administración de MYLOTARG®
Hepatotoxicidad: La hepatotoxicidad, incluyendo la enfermedad hepática severa veno-oclusiva (SVO), ha sido reportada en asociación con el uso de MYLOTARG® como un agente único, como parte de un régimen de quimioterapia combinada, y en pacientes sin antecedentes de enfermedad hepática o trasplante de células madre hematopoyéticas (Stem cell HSCT) (ver Reacciones adversas). Los pacientes que reciben MYLOTARG® antes o después de HSCT, los pacientes con enfermedad hepática implícita o función hepática anormal, y los pacientes recibiendo MYLOTARG® en combinación con otra quimioterapia pueden tener un riesgo más alto de desarrollar SVO. Se ha reportado la muerte por falla hepática y por SVO en pacientes que recibían MYLOTARG®. Los médicos deben supervisar cuidadosamente sus pacientes por síntomas de hepatotoxicidad, particularmente la enfermedad SVO. Estos síntomas pueden incluir: ganancia rápida de peso, dolor en el cuadrante superior derecho, hepatomegalia, ascitis, elevación de la bilirrubina y/o enzimas del hígado. Sin embargo, con una supervisión cuidadosa no se pueden identificar todos los pacientes de riesgo o prevenir las complicaciones de la hepatotoxicidad.
General MYLOTARG® debe ser administrado bajo la supervisión de médicos con experiencia en el tratamiento de la leucemia aguda y en instalaciones equipadas para supervisar y tratar a los pacientes con leucemia.
No existen estudios clínicos controlados que demuestren la eficacia y seguridad del uso de MYLOTARG® en combinación con otros agentes quimioterapéuticos. Por lo tanto, MYLOTARG® debe ser solamente usado como una quimioterapia de agente único y no en regímenes de quimioterapia de combinación por fuera de los estudios clínicos.
Mielosupresión: Puede ocurrir mioelosupresión severa en todos los pacientes que reciben la dosis recomendada de este agente. Se requiere una cuidadosa supervisión hematológica. Las infecciones sistémicas deberían ser tratadas.
Reacciones de hipersensibilidad incluyendo anafilaxis, reacciones a la infusión, eventos pulmonares: La administración de MYLOTARG® puede tener como resultado reacciones severas de hipersensibilidad (incluyendo anafilaxis), y otras reacciones relacionadas con la infusión que pueden concluir en eventos severos pulmonares. Raras veces han sido fatales las reacciones de hipersensibilidad y los eventos pulmonares.
En la mayoría de los casos, los síntomas ocurridos en relación con la infusión o dentro de las 24 horas a la administración de MYLOTARG® se han resuelto. La infusión de MYLOTARG® (R) debe ser interrumpida en pacientes que experimenten disnea o hipotensión clínicamente significativa. Los pacientes deben ser supervisados hasta que los signos y síntomas sean completamente resueltos. La descontinuación del tratamiento con MYLOTARG® debe ser revisado en pacientes que desarrollan anafilaxis, edema pulmonar o síndrome de peligro respiratorio agudo. Como los pacientes con alto recuento de blastos periféricos pueden tener un riesgo más grande de eventos pulmonares y síndrome de lisis tumoral, los médicos deben considerar la leucorreducción con hidroxiurea o leucoferesis para reducir el recuento de los glóbulos blancos periféricos por debajo de 30.000/µL antes de la administración de MYLOTARG®.
Reacciones a la infusión: MYLOTARG® puede producir un complejo de síntomas posinfusión de fiebre y escalofríos, y menos comúnmente de hipotensión y disnea que pueden ocurrir dentro de las 24 horas después de su administración. Los efectos adversos relacionados con la infusión de grado 3 ó 4 no hematológicos incluyen escalofríos, fiebre, hipotensión, hipertensión, hiperglicemia, hipoxia y disnea. La mayoría de los pacientes recibieron las siguientes medicaciones profilácticas antes de la administración: difenhidramina 50 mg vía oral y acetaminofén 650-1.000 mg vía oral después de esto, dos dosis adicionales de acetaminofén 650-1.000 mg vía oral, una cada cuatro horas en caso necesario. Los signos vitales deben ser supervisados durante la infusión y durante las cuatro horas siguientes a la infusión.
En estudios clínicos, generalmente estos síntomas ocurrieron después de finalizar la infusión intravenosa de dos horas y desaparecen después de dos a cuatro horas con una terapia de soporte con acetaminofén, difenhidramina y fluidos IV. Se observaron muy pocos eventos relacionados con la infusión después de la segunda dosis.
Eventos pulmonares: Rara vez se han reportado eventos severos pulmonares que lleven a la muerte con el uso de MYLOTARG® en el ambiente del posmercadeo. Los signos, síntomas y hallazgos clínicos incluyen disnea, infiltrados pulmonares, efusiones pleurales, edema pulmonar no cardiogénico, insuficiencia pulmonar e hipoxia, y síndrome de dificultad respiratoria aguda. Estos eventos ocurren como secuelas de la reacción a la infusión; los pacientes con recuento de glóbulos blancos>30.000/µL pueden estar en un riesgo aumentado. Los médicos deben considerar la leucorreducción con hidroxiurea o leucoferesis para reducir el recuento de glóbulos blancos periféricos a menos de 30.000/µL antes de la administración de MYLOTARG®. Los pacientes con enfermedad pulmonar intrínseca sintomática pueden también estar en un mayor riesgo de presentar reacciones pulmonares severas.
Hepatotoxicidad: La hepatotoxicidad, incluyendo enfermedad hepática severa y en enfermedad venooclusiva (SVO) severa, ha sido reportada en asociación con el uso de MYLOTARG® como un agente único, como parte de un régimen de quimioterapia combinada, y en pacientes sin antecedentes de enfermedad hepática o trasplante de células madre HSCT (Stem cell) (ver sección Reacciones adversas). Los pacientes que reciben MYLOTARG® antes o después de HSCT, pacientes con enfermedad hepática implícita o función hepática anormal, y pacientes recibiendo MYLOTARG® en combinación con otra quimioterapia pueden estar en gran riesgo de desarrollar SVO. Se han reportado muertes por falla hepática y de SVO en pacientes recibiendo MYLOTARG®. Los médicos deben supervisar cuidadosamente a sus pacientes por síntomas de hepatotoxicidad, particularmente SVO. Estos síntomas pueden incluir: ganancia rápida de peso, dolor en el cuadrante superior derecho, hepatomegalia, ascitis, elevaciones en la bilirrubina y/o de las enzimas hepáticas. Sin embargo, la supervisión cuidadosa puede no identificar todos los pacientes con riesgo o prevenir las complicaciones de la hepatotoxicidad.
Uso en pacientes con deterioro hepático: MYLOTARG® no ha sido estudiado en pacientes con bilirrubina >2 mg/dL. Se deben tomar precauciones extra cuando se administre MYLOTARG® en pacientes con deterioro hepático (ver sección Reacciones adversas).
El Síndrome de Lisis Tumoral: El Síndrome de Lisis Tumoral (SLT) puede ser una consecuencia del tratamiento de la leucemia con cualquier agente quimioterapéutico incluyendo MYLOTARG®. Se ha reportado falla renal secundaria a SLT con el uso de MYLOTARG®. Se deben tomar medidas apropiadas (ej.: hidratación y alopurinol) para prevenir la hiperuricemia. Los médicos deben considerar la leucorreducción con hidroxiurea o leucoferesis para reducir el recuento de glóbulos blancos periféricos por debajo de 30.000/µL antes de la administración de MYLOTARG®.
Embarazo: MYLOTARG® puede causar daño fetal cuando se administra a mujeres embarazadas. No existen estudios adecuados ni bien controlados en mujeres embarazadas. Si MYLOTARG® es usado durante el embarazo, o si la paciente se embaraza mientras esté tomándolo, la paciente debe ser advertida de los posibles riesgos para el feto. Las mujeres que están en edad reproductiva deben ser aconsejadas de evitar el embarazo mientras estén recibiendo tratamiento con MYLOTARG®.
PRECAUCIONES
Exámenes de laboratorio: Se debe supervisar los electrolitos, pruebas de la función hepática, recuento sanguíneo completo (RSC) y recuento de plaquetas durante la terapia con MYLOTARG®.
Uso en pacientes con desorden renal: Los pacientes con desorden renal no fueron estudiados.
EMBARAZO: MYLOTARG® puede causar daño fetal cuando se administra a una mujer embarazada. El tratamiento diario con gemtuzumab ozogamicina a ratas embarazadas durante la organogénesis produjo disminuciones del peso fetal en asociación con la disminución de la osificación esquelética fetal relacionadas con la dosis empezando en 0.025 mg/kg/día. Dosis de 0.060 mg/kg/día (aproximadamente 0.04 veces la dosis única humana recomendada sobre una base de mg/m2) produjo un aumento de la mortalidad del embrión fetal (aumentó el número de reabsorción y disminuyó el número de fetos vivos por camada). Manifiestas alteraciones externas, viscerales y esqueléticas a la dosis de 0.060 mg/kg/día incluyeron malformaciones digitales (ectrodactilia, braquidactilia), en uno o en ambas patas posteriores ausencia del arco aórtico, costillas hundidas, anomalías de los huesos largos en los miembros delanteros (húmero corto/grueso, radio y cúbito deformados, y cúbito corto/grueso), escápula deformada, ausencia de centro vertebral, y vértebras esternales fusionadas. Esta dosis también se asoció con toxicidad maternal (disminución de la ganancia de peso, disminución del consumo de alimento). No hay estudios adecuados y bien controlados en mujeres embarazadas. Si MYLOTARG® es usado en el embarazo, o si la paciente se embaraza mientras lo toma, la paciente debe ser advertida del riesgo potencial para el feto. Las mujeres en edad reproductiva deben ser advertidas de no quedar embarazadas mientras reciban el tratamiento con MYLOTARG®.
LACTANCIA: No se sabe si MYLOTARG® se excreta en la leche humana. Debido a que muchas drogas, incluyendo las inmunoglobulinas, son excretadas en la leche humana y a causa del potencial de reacciones adversas serias en infantes lactantes relacionadas con MYLOTARG® se debe tomar una decisión con respecto a si se discontinúa la lactancia o la droga, teniendo en cuenta la importancia de la droga para la madre.
USO PEDIATRICO: Todavía no han sido estudiadas la seguridad y efectividad de MYLOTARG® en pacientes pediátricos.
INTERACCIONES: No se han realizado estudios formales sobre las interacciones medicamentosas con MYLOTARG®.
INTERFERENCIA CON LOS ANALISIS DE LABORATORIO Y DE DIAGNOSTICO: No se conoce que MYLOTARG® interfiera cualquier prueba diagnóstica de rutina.
REACCIONES ADVERSAS: En los estudios clínicos para el registro, MYLOTARG® se administró a 142 pacientes con recaída de LMA (Leucemia Mieloide Aguda) a 9 mg/m2. MYLOTARG® se suministró en general como dos infusiones intravenosas separadas por 14 días.
Eventos agudos relacionados con la infusión

Estos síntomas ocurrieron generalmente después de terminar la infusión intravenosa de dos horas y se resolvieron después de dos a cuatro horas con una terapia de soporte compuesta por acetaminofén, difenhidramina y fluidos IV (ver sección Advertencias especiales). Se observaron muy pocos eventos relacionados con la infusión después de la segunda dosis.
Formación de anticuerpos: No se detectaron anticuerpos contra el gemtuzumab ozogamicina en un total de 142 pacientes en los estudios clínicos Fase 2. Dos pacientes en un estudio Fase 1 desarrollaron títulos anticuerpo contra la porción caliqueamicina/porción ligada de caliqueamicina de gemtuzumab ozogamicina después de tres dosis. Un paciente experimentó fiebre transitoria, hipotensión y disnea; el otro paciente no tuvo síntomas clínicos. Ningún paciente desarrolló respuesta de anticuerpo a la porción anticuerpo hP67.6 del MYLOTARG®.
Mielosupresión: La mielosupresión severa es la principal toxicidad asociada con MYLOTARG®. Durante la fase de tratamiento, 137/140 (98%) pacientes experimentaron neutropenia Grado 3 o Grado 4. Los pacientes que respondieron (RC: remisión completa y Rcp: remisión parcial), recobraron su recuento absoluto de neutrófilos absolutos a 500/µL con una mediana de 40.5 días después de la primera dosis de MYLOTARG®.
Anemia, trombocitopenia: Durante la fase de tratamiento, 139/141 (99%) pacientes experimentaron trombocitopenia Grado 3 o Grado 4. Los pacientes que respondieron (pacientes RC y RCp; ver sección Farmacodinámica, eficacia clínica - General para la explicación de las abreviaciones) recobraron los recuentos de plaquetas a 25.000/µL con una mediana de 39 días después de la primera dosis de MYLOTARG®. 66/141 (47%) pacientes experimentaron anemia Grado 3 o Grado 4.
Infección: Durante la fase de tratamiento, 40/142 (28%) pacientes experimentaron infecciones Grado 3 o Grado 4, incluyendo infecciones oportunistas. Los eventos adversos emergentes de las infecciones más frecuentes Grado 3 o Grado 4 relacionadas con el tratamiento fueron sepsis (16%) y neumonía (7%). Se reportó infección por herpes simple en 22% de los pacientes.
Hemorragia: Durante la fase de tratamiento, 21/142 (15%) pacientes experimentaron hemorragia Grado 3 o Grado 4. El evento adverso severo más frecuente que emergió del tratamiento fue epistaxis (3%). También hubo reportes de hemorragia cerebral (2%), coagulación intravascular diseminada (2%), hemorragia intracraneal (2%) y hematuria (1%).
Transfusiones: Durante la fase de tratamiento se requirieron más transfusiones para los pacientes no respondedores (NR) y en los de remisión parcial (RCp) comparados con los pacientes de remisión completa (RC) (ver sección Farmacodinámica, eficacia clínica - General para la explicación de las abreviaciones) (Tabla 2):

Mucositis: Se reportó un total de 50/142 (35%) pacientes con eventos adversos que emergieron del tratamiento consistentes en mucositis oral y estomatitis. Durante la fase de tratamiento, 5/142 (4%) pacientes experimentaron una estomatitis/mucositis Grado 3 ó 4 después de la primera dosis. Los eventos de la mucositis en el resto de los 45/142 (32%) pacientes se categorizaron como Grado 1 ó 2.
Hepatotoxicidad: Las anormalidades de la función hepática fueron transitorias y generalmente reversibles. En estudios clínicos, 33/141 (23%) pacientes experimentaron hiperbilirrubinemia Grado 3 o Grado 4. Nueve por ciento (12/141) de pacientes experimentaron anormalidades Grado 3 o Grado 4 en los niveles de ALT, y 24/141 (17%) pacientes experimentaron anormalidades Grado 3 o Grado 4 en los niveles de AST. Trece pacientes tuvieron elevaciones concurrentes de transaminasas (grado 3 a 4) y bilirrubina. Un paciente murió por falla hepática relacionada al síndrome de lisis tumoral y falla orgánica multisistémica 22 días después del tratamiento. Otro paciente murió después de un episodio de ictericia persistente y hepatoesplenomegalia 156 días después del tratamiento. Entre 27 pacientes que recibieron trasplante de células madre hematopoyéticas siguiendo la administración de MYLOTARG®, tres (2 NR y 1 RC; ver sección Farmacodinámica, eficacia clínica - General para la explicación de las abreviaciones) murieron de enfermedad hepática veno-oclusiva (VO) 22 a 35 días después del trasplante.
Piel: Ningún paciente experimentó alopecia. Se reportó un brote no específico en 22% de los pacientes.
Cursos repetidos: Cinco pacientes han recibido más de un curso de MYLOTARG®, cuatro de estos pacientes 9 mg/m2. El perfil de evento adverso para pacientes retratados fue similar al que siguió su tratamiento inicial. Uno de los pacientes a quien se le repitió la dosis que estaba en un estudio Fase 1 y recibió un primer curso de tres dosis de 1 mg/m2 y dos dosis de un segundo curso de 6 mg/m2. Este paciente fue descontinuado de la administración de una dosis posterior como resultado de una respuesta inmune a la porción caliqueamicina/porción ligada a la caliqueamicin de gemtuzumab ozogamicina. Los otros cuatro pacientes vueltos a tratar no experimentaron ninguna respuesta inmune.
Relación de la dosis con los eventos adversos: Los datos sobre la relación de la dosis fueron generados de un estudio pequeño de escalonamiento de dosis. El evento clínico adverso más común observado en este estudio fue un complejo de síntomas relacionados con la infusión de fiebre y escalofríos. En general, la severidad de la fiebre, pero no los escalofríos, aumentó a medida que el nivel de la dosis aumentaba. Sólo un nivel de dosis de MYLOTARG® fue estudiado en los estudios clínicos de Fase 2 en LMA (Leucemia Mieloide Aguda) en recaída.
Eventos adversos emergentes del tratamiento (EAET): Los eventos adversos emergentes del tratamiento (Grados 1-4) que ocurrieron en > 10% de los pacientes sin tener en cuenta la causalidad se listan en la Tabla 3.

a. No incluye cambios en los valores de laboratorio reportados como eventos adversos para eventos incluidos en la escala de toxicidad común del Instituto Nacional de Cáncer en los Estados Unidos (INC).
b. >10% del límite especifica el umbral de porcentaje mínimo de por lo menos una columna para que el evento aparezca en la tabla.
c. Incluye estertores, ronquido y cambios en los sonidos de la respiración
d. Porcentajes para eventos adversos específicos para el sexo están basados en el número de pacientes del sexo relevante.
Eventos adversos emergentes del tratamiento con una severidad Grado 3 ó 4 se listan en el Tabla 4.
Tabla 4.
Porcentaje (%) de pacientes reportando eventos
adversos emergentes del tratamiento severos en
la escala grado 3 ó 4 (Incidencia > 5%b)

a. No incluye cambios en los valores del laboratorio reportados como eventos adversos incluidos en la escala de toxicidad común del Instituto Nacional de Cáncer de los Estados Unidos (INC).
b. >5% límite específico del límite especifica el umbral de porcentaje mínimo de por lo menos una columna para que el evento aparezca en la tabla.
Anormalidades de laboratorio clínicamente importantes en el laboratorio con una severidad Grado 3 ó 4 se listan en la Tabla 5.
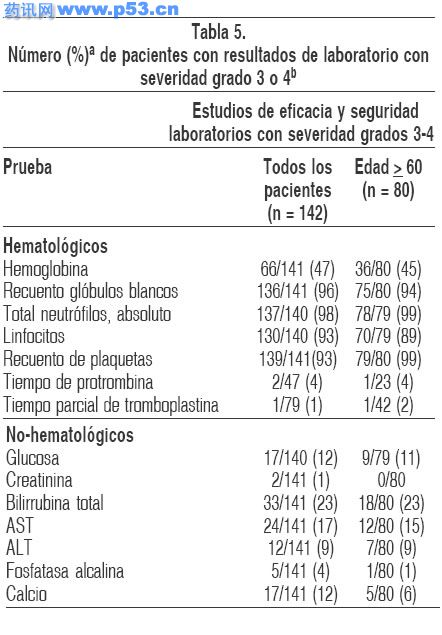
a. El porcentaje está basado en el número de pacientes que recibieron una prueba de un laboratorio en particular durante el estudio como se indica para cada prueba.
b. La severidad definida por la escala de toxicidad común del Instituto Nacional de Cancerología de los Estados Unidos (INC) versión 1.
Las diferencias en los eventos adversos emergentes del tratamiento entre pacientes 60 años y aquellos pacientes >60 no se consideraron clínicamente importantes. Los parámetros de laboratorio asociados con disfunción hepática (ej.: niveles elevados de bilirrubina, AST, y ALT) fueron observados más consistentemente en pacientes > 60 años que en aquellos <60 años.
Las diferencias en los eventos adversos emergentes del tratamiento entre pacientes hombres y mujeres no se consideraron clínicamente importantes.
Otras experiencias clínicas: Se han reportado casos adicionales de la enfermedad venooclusiva VO en experiencias posmercadeo y otros estudios clínicos, algunos asociados con el uso de otros agentes quimioterapéuticos, bajo enfermedad hepática implícita/función anormal del hígado, o historia de HSCT anterior o subsecuente. También han sido reportados en asociación con el uso de MYLOTARG® falla renal secundaria a SLT (síndrome de lisis tumoral), reacciones de hipersensibilidad, anafilaxis, eventos pulmonares y hemorragia gastrointestinal (ver sección Advertencias especiales).
SOBREDOSIS: En experiencias clínicas no se han reportado casos de sobredosis con MYLOTARG®. No se analizaron dosis únicas por encima de 9 mg/m2 en adultos. Cuando se administró una dosis única de MYLOTARG® a animales, se observó una mortalidad en ratas a la dosis de 2 mg/kg (aproximadamente 1.3 veces la dosis humana recomendada en una base de mg/m2), y en simios machos a la dosis de 4.5 mg/kg (aproximadamente seis veces la dosis humana recomendada en una base de mg/m2).
Las medidas de soporte general deben seguirse en casos de sobredosis. Se deben supervisar cuidadosamente la presión arterial y el recuento sanguíneo. El gemtuzumab ozogamicina no es dializable.
MECANISMO DE ACCION: La gemtuzumab ozogamicina se enlaza al antígeno CD33. Este antígeno se expresa en la superficie de los blastos leucémicos en más del 80% de los pacientes con leucemia mieloidea aguda (LMA). El CD33 se expresa también sobre las células que forman las colonias de leucemia mieloide, incluyendo precursores clonogénicos, pero no se expresa en las células madre hematopoyéticas pluripotentes o sobre las células no hematopoyéticas.
MYLOTARG® está dirigido contra el antígeno CD33 expresado por las células hematopoyéticas. La unión de la porción anticuerpo del anti-CD33 de MYLOTARG® con el antígeno CD33 resulta en la formación de un complejo que es internalizado. Después de la internalización, el derivado caliqueamicina es liberado dentro de los lisosomas de la célula mieloide. El derivado liberado de la caliqueamicina se enlaza al ADN en el canal menor resultando en que la doble hélice de ADN se rompe y la célula muere.
El gemtuzumab ozogamicina es citotóxico a la línea celular leucémica humana positiva de HL-60 del CD33. El gemtuzumab ozogamicina produce inhibición significativa de la formación de colonias en cultivos de las células leucémicas adultas de la médula ósea. El efecto citotóxico sobre los precursores mieloides normales lleva a una mielosupresión sustancial, pero esto es reversible porque las células madre hematopoyéticas pluripotentes son de reserva. En estudios preclínicos en animales, el gemtuzumab ozogamicina demostró efectos antitumorales en el tumor del trasplante heterólogo humano de la leucemia promielocítica del HL-60 en el ratón sin timo.
FARMACODINAMICA, EFICACIA CLINICA
General: La eficacia y seguridad de MYLOTARG® como un agente único han sido evaluadas en 142 pacientes en tres estudios abiertos únicos en pacientes con LMA positiva para CD33 en la primera recaída. Los estudios incluyeron 65, 40 y 37 pacientes. En los estudios 1 y 2 los pacientes eran > 18 años con una duración de la primera remisión de por lo menos seis meses. En el estudio 3, sólo pacientes > 60 años fueron reclutados y su primera remisión había terminado por lo menos hacía tres meses. Los pacientes con leucemia secundaria o recuentos de glóbulos blancos (CGB) de >30.000/µL fueron excluidos. Algunos pacientes eran leucorreducidos con hidroxiurea o leucoferesis para bajar el recuento de CGB por debajo de 30.000/µL con el fin de minimizar el riesgo de síndrome de lisis tumoral. El curso del tratamiento incluyó dos dosis de 9 mg/m2 separadas por un intervalo de 14 días y un seguimiento de 28 días después de la última dosis. Aunque dosis más pequeñas habían producido respuestas en estudios previos, la dosis de 9 mg/m2 fue escogida porque se esperaba saturar todos los sitios CD33 sin tener en cuenta el impacto leucémico. Un total de 80 pacientes tenían 60 años y más. El objetivo primario de los tres estudios clínicos fue la tasa de remisión completa (RC), que fue definida como:
- Ausencia de blastos leucémicos de la sangre periférica;
- <5% de blastos en la médula ósea, según la medición de los estudios morfológicos;
- Hemoglobina (Hgb) > 9 g/dL, plaquetas > 100.000/µL, recuento absoluto de neutrófilos (CNA) > 1.500/µL, y
- No dependencia de transfusión de glóbulos rojos y plaquetas (sin transfusión de glóbulos rojos por dos semanas; sin transfusión de plaquetas por una semana).
En adición a RC, una segunda categoría de respuesta, RCp, se definió como aquellos pacientes que satisfacen la definición de RC, incluyendo no dependencia de transfusión de plaquetas, con excepción de la recuperación de plaquetas > 100.000/µL. Esta categoría fue añadida porque MYLOTARG® parece demorar la recuperación de plaquetas en algunos pacientes. La mayoría de estos pacientes (18/19) alcanzaron recuento de plaquetas de por lo menos 25.000/µL y cerca de dos terceras partes (13/19) alcanzaron recuento de plaquetas de por lo menos 50.000/µL, antes de que cualquier terapia adicional fuera administrada. Aún no es claro si las respuestas RC o RCp son clínicamente equivalentes; pero la sobrevivencia en los dos grupos pareció similar.
Todos los pacientes fueron premedicados con acetaminofén 650-1.000 mg y difenhidramina 50 mg para disminuir los síntomas agudos relacionados con la infusión. Los factores de crecimiento y citoquinas no fueron permitidos. No se especificó el uso de antibióticos profilácticos.
Tasa de respuesta: La tasa total de respuesta (TTR) para los tres estudios agrupados fue 30% (42/142) consistiendo de 16% (23/142) de pacientes con RC y 13% (19/142) de pacientes con RCp. El tiempo promedio para la remisión fue 60 días tanto para RC como para RCp. Las tasas de remisión en los estudios individuales se muestran en la Tabla 6.
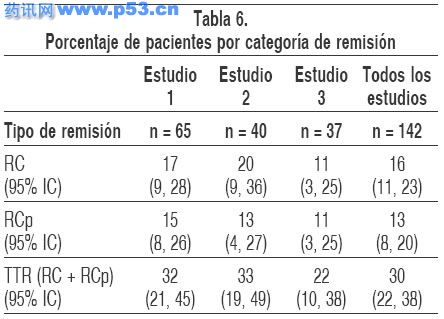
a. Pacientes 60 años de edad o mayores.
Dos de las determinantes más importantes de respuesta siguiendo la recaída son la edad y la duración de la primera remisión. Las tasas de remisión por categoría de pronóstico se muestran en la Tabla 7; el impacto de la edad y duración de la primera remisión en estos pacientes fueron mínimos:
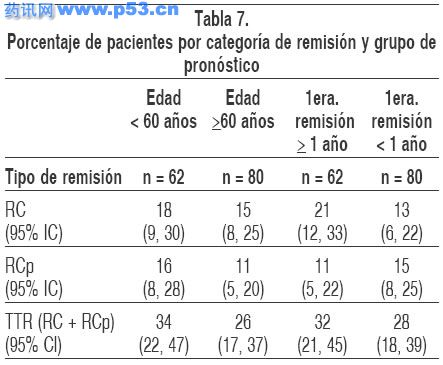
Entre los pacientes <60 años la tasa total de respuesta fue 34%; entre los pacientes >60 años de edad la tasa total de respuesta fue 26%. Las tasas totales de respuesta fueron similares para mujeres y hombres; 31% mujeres y 29% hombres alcanzaron la remisión.
La mayoría de los pacientes (94%) en los estudios clínicos fase 2 fueron blancos, solo 6% no eran blancos. Todos los 42 pacientes que respondieron eran blancos.
Sobrevida libre de recaída: La sobrevida libre de recaída fue calculada de los datos de la terapia inicial (Tabla 8).

a. Número de meses después de alcanzar RC o RCp.
b. Los datos son limitados por la fecha de corte, el primer evento ocurrió dentro de los 0.83 meses para RCp y en 0.5 meses para la TTR.
c. Seis pacientes TTR (1 RC y 5 RCp) tuvieron sobrevivencia libre de recaída >12 meses.
Sobrevida total: El promedio de sobrevida total para los 142 pacientes fue 5.9 meses y 55/142 pacientes estaban vivos a la fecha de corte de datos.
Terapia posremisión: Quince (15/42, 36%) pacientes con TTR (8 RC y 7 RCp) recibieron trasplante de células madre hematopoyéticas. La supervivencia de estos 15 pacientes osciló de 3.5 a 26.9 meses a la fecha de corte de datos. Nueve pacientes TTR (4 RC y 5 RCp) tuvieron una sobrevida total > 12 meses a la fecha de corte de datos.
Cursos repetidos: Cinco pacientes habían recibido un segundo curso de tratamiento de MYLOTARG® en estudios clínicos. Estos pacientes fueron inicialmente tratados con MYLOTARG®, alcanzaron remisión, y subsecuentemente recayeron. Uno de estos pacientes (> 60 años) alcanzó una segunda RC después de recibir el segundo curso de MYLOTARG®. Se observó una mielosupresión severa prolongada en cuatro pacientes que estaban recibiendo una tercera dosis.
Revisión general de los datos clínicos: Los datos disponibles del estudio simple no suministraron comparaciones válidas con varios regímenes citotóxicos que habían sido usados en leucemia mieloide aguda en recaída. Las tasas de respuesta están en el rango de tasas reportadas con tales regímenes sólo si las respuestas RCp están incluidas. Sin embargo, el tratamiento con MYLOTARG® puede suministrar respuestas, incluyendo algunas de duración razonable. Los datos respaldan su uso en pacientes para quienes los regímenes agresivos citotóxicos podrían considerarse no aceptables, tales como muchos pacientes de 60 años y mayores.
FARMACOCINETICA: Después de la administración de la primera dosis recomendada de 9 mg/m2 de gemtuzumab ozogamicina, administrada como una infusión de 2 horas, la vida media de eliminación de la caliqueamicina total y no conjugada fue de alrededor de 45 y 100 horas respectivamente. Después de la segunda dosis de 9 mg/m2, la vida media de la caliqueamicina total aumentó alrededor de 60 horas y el área bajo la curva de concentración/tiempo (AUC) fue de alrededor de dos veces del periodo de la primera dosis. La farmacocinética de la caliqueamicina no conjugada no pareció cambiar del periodo uno al segundo periodo. Los estudios metabólicos indican la liberación hidrolítica del derivado caliqueamicina del gemtuzumab ozogamicina. Se encontraron muchos metabolitos de este derivado después de la incubación in vitro de gemtuzumab ozogamicina en los microsomas hepáticos humanos y citosol, y las célula leucémicas promielocíticas HL60. No se han efectuado estudios metabólicos caracterizando las posibles isoenzimas involucradas en la vía metabólica de MYLOTARG®.
DATOS DE SEGURIDAD PRECLINICA
Carcinogénesis, mutagénesis, deterioro de la fertilidad: No se han efectuado estudios a largo plazo en animales para evaluar el potencial carcinogénico de MYLOTARG®. El gemtuzumab ozogamicina fue clastogénico en el ratón en la prueba micronuclear in vivo. Este resultado positivo es consistente con la habilidad conocida de la caliqueamicina de causar rupturas de la doble hélice del ADN. El gemtuzumab ozogamicina afectó adversamente la fertilidad del macho, pero no de la hembra en ratas. Siguiendo la administración diaria de gemtuzumab ozogamicina a ratas machos durante 28 días a dosis de 0.02 a 0.16 mg/kg/día (aproximadamente 0.01 a 0.11 veces la dosis humana en una base de mg/m2), el gemtuzumab ozogamicina causó: disminución de las tasas de fertilidad, recuentos de esperma epididimarios, y de la movilidad de los espermatozoides; aumentó la incidencia de anormalidades en los espermatozoides; y evidencia microscópica de disminución en el recuento de espermatogonia y espermatocitos. Estos hallazgos no se resolvieron siguiendo un periodo de recuperación de nueve semanas.
COMPATIBILIDADES, INCOMPATIBILIDADES: Para reconstitución se usa agua estéril para inyección. Se usa una solución de cloruro de sodio 0.9% para dilución de la solución reconstituida.
MANIPULACION
General: Se deben emplear los procedimientos institucionales para la manipulación de las drogas anticancerosas.
Instrucciones para uso, manipulación y eliminación: Los viales sólo para un solo uso. A través de toda la manipulación de MYLOTARG® deben emplearse estrictas técnicas asépticas ya que el producto no tiene un agente bacteriostático o preservativo.
MYLOTARG® es sensible a la luz y debe protegerse de los rayos de sol directos o indirectos y de la luz fluorescente sin filtro durante la preparación y administración de la infusión (usando una bolsa protectora de las radiaciones [UV] sobre la bolsa IV durante la infusión). Toda la preparación debe efectuarse en una cabina biológicamente segura con la luz fluorescente protegida con una armadura protectora.
Instrucciones para la reconstitución: Reconstituir los componentes de cada vial con 5 mL de agua estéril para inyección, usando jeringas estériles. Agitar suavemente cada vial. Cada vial debe ser visualmente inspeccionado para verificar la completa disolución del medicamento. La concentración final del medicamento reconstituido en el vial es de 1 mg/mL.
La solución de MYLOTARG® reconstituida debe ser inspeccionada visualmente para descartar presencia de partículas y decoloración, antes de ser transferida a la jeringa. Adicionalmente, la solución de la mezcla debe ser inspeccionada visualmente para descartar presencia de partículas y decoloración.
La solución del medicamento en el vial, transferida a la jeringa, o la bolsa de la mezcla puede parecer turbia por dispersión normal de la luz de la proteína.
Ver también la sección para Almacenamiento información sobre Dilución, almacenamiento e infusión.
Instrucciones para dilución: Prepare la mezcla correspondiente a una dosis de MYLOTARG® 9 mg/m2 inyectando la solución reconstituida en 100 mL de una solución de cloruro de sodio para inyección al 0,9% en una bolsa de cloruro polivinílico (PVC) o de etileno/polipropileno copolímero (no PVC) cubierta por un protector de luz ultravioleta (UV). MYLOTARG® sólo puede ser diluido con una solución de cloruro de sodio 0,9%. NO DILUIR CON CUALQUIER OTRA SOLUCION DE ELECTROLITOS, DEXTROSA AL 5%, NI MEZCLAR CON OTROS MEDICAMENTOS.
Ver también la sección
Almacenamiento para Información sobre dilución, almacenamiento e infusión.
ALMACENAMIENTO
Antes de la reconstitución: MYLOTARG® debe ser almacenado refrigerado (2 °C a 8 °C, 36 °F a 46 °F) y protegido de la luz.
Después de diluido: Seguir las instrucciones antes mencionadas para la reconstitución, dilución y administración. Ver las siguientes tablas para las condiciones de almacenamiento e intervalos para la reconstitución, dilución y administración.

a Tiempo máximo total de almacenamiento desde la reconstitución del producto hasta la infusión completa.
MANEJO DE DESECHOS: Deben emplearse los procedimientos prescritos para la eliminación de los desechos tóxicos de los medicamentos anticancerígenos.
PRESENTACION COMERCIAL Y CONDICION DE VENTA: MYLOTARG®, caja plegable de cartón con 1 vial de unidosis de MYLOTARG® color ámbar, que contiene polvo liofilizado estéril, libre de preservantes con 5 mg de gemtuzumab ozogamicina y 1 frasco con 5 mL de diluyente para MYLOTARG® (agua estéril para inyección, USP) (Reg. San. INVIMA 2002M-0000999). Venta con fórmula médica. | 
