|
ORENCIA(abatacept) HIGHLIGHTS OF PRESCRIBING INFORMATION
FULL PRESCRIBING INFORMATION 1 INDICATIONS AND USAGE 1.1 Adult Rheumatoid Arthritis (RA) ORENCIA® is indicated for reducing signs and symptoms, inducing major clinical response, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis. ORENCIA may be used as monotherapy or concomitantly with disease-modifying antirheumatic drugs (DMARDs) other than tumor necrosis factor (TNF) antagonists. 1.2 Juvenile Idiopathic Arthritis ORENCIA is indicated for reducing signs and symptoms in pediatric patients 6 years of age and older with moderately to severely active polyarticular juvenile idiopathic arthritis. ORENCIA may be used as monotherapy or concomitantly with methotrexate (MTX). 1.3 Important Limitations of Use ORENCIA should not be administered concomitantly with TNF antagonists. ORENCIA is not recommended for use concomitantly with other biologic rheumatoid arthritis (RA) therapy, such as anakinra. 2 DOSAGE AND ADMINISTRATION 2.1 Adult Rheumatoid Arthritis For adult patients with RA, ORENCIA may be administered as an intravenous infusion or a subcutaneous injection. ORENCIA may be used as monotherapy or concomitantly with DMARDs other than TNF antagonists. For pediatric juvenile idiopathic arthritis, a dose calculated based on each patient's body weight is used [see Dosage and Administration (2.2)]. Intravenous Dosing Regimen ORENCIA intravenous should be administered as a 30-minute intravenous infusion utilizing the weight range-based dosing specified in Table 1. Following the initial intravenous administration, an intravenous infusion should be given at 2 and 4 weeks after the first infusion and every 4 weeks thereafter.
Following a single intravenous loading dose (as per body weight categories listed in Table 1), the first 125 mg subcutaneous injection of ORENCIA should be given within a day, followed by 125 mg subcutaneous injections once weekly. Patients who are unable to receive an infusion may initiate weekly injections of subcutaneous ORENCIA without an intravenous loading dose. Patients transitioning from ORENCIA intravenous therapy to subcutaneous administration should administer the first subcutaneous dose instead of the next scheduled intravenous dose. 2.2 Juvenile Idiopathic Arthritis The recommended dose of ORENCIA for patients 6 to 17 years of age with juvenile idiopathic arthritis who weighless than 75 kg is 10 mg/kg intravenously calculated based on the patient’s body weight at each administration. Pediatric patients weighing 75 kg or more should be administered ORENCIA following the adult intravenous dosing regimen, not to exceed a maximum dose of 1000 mg. ORENCIA should be administered as a 30-minute intravenous infusion. Following the initial administration, ORENCIA should be given at 2 and 4 weeks after the first infusion and every 4 weeks thereafter. Any unused portions in the vials must be immediately discarded. 2.3 Preparation and Administration Instructions for Intravenous Infusion Use aseptic technique. ORENCIA is provided as a lyophilized powder in preservative-free, single-use vials. Each ORENCIA vial provides 250 mg of abatacept for administration. The ORENCIA powder in each vial must be reconstituted with 10 mL of Sterile Water for Injection, USP, using only the silicone-free disposable syringe provided with each vial and an 18- to 21-gauge needle. After reconstitution, the concentration of abatacept in the vial will be 25 mg/mL. If the ORENCIA powder is accidentally reconstituted using a siliconized syringe, the solution may develop a few translucent particles. Discard any solutions prepared using siliconized syringes. If the silicone-free disposable syringe is dropped or becomes contaminated, use a new silicone-free disposable syringe from inventory. For information on obtaining additional silicone-free disposable syringes, contact Bristol-Myers Squibb 1-800-ORENCIA.
2.4 General Considerations for Subcutaneous Administration ORENCIA Injection, 125 mg/syringe is not intended for intravenous infusion. ORENCIA Injection is intended for use under the guidance of a physician or healthcare practitioner. After proper training in subcutaneous injection technique, a patient may self-inject with ORENCIA if a physician/healthcare practitioner determines that it is appropriate. Patients should be instructed to follow the directions provided in the Instructions for Use for additional details on medication administration. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use ORENCIA prefilled syringes exhibiting particulate matter or discoloration. ORENCIA should be clear and colorless to pale yellow. Patients using ORENCIA for subcutaneous administration should be instructed to inject the full amount in the syringe (1 mL), which provides 125 mg of ORENCIA, according to the directions provided in the Instructions for Use. Injection sites should be rotated and injections should never be given into areas where the skin is tender, bruised, red, or hard. 3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS None. 5 WARNINGS AND PRECAUTIONS 5.1 Concomitant Use with TNF Antagonists In controlled clinical trials in patients with adult RA, patients receiving concomitant intravenous ORENCIA and TNF antagonist therapy experienced more infections (63%) and serious infections (4.4%) compared to patients treated with only TNF antagonists (43% and 0.8%, respectively) [see Adverse Reactions (6.1)]. These trials failed to demonstrate an important enhancement of efficacy with concomitant administration of ORENCIA with TNF antagonist; therefore, concurrent therapy with ORENCIA and a TNF antagonist is not recommended. While transitioning from TNF antagonist therapy to ORENCIA therapy, patients should be monitored for signs of infection. 5.2 Hypersensitivity Of 2688 patients with adult RA treated with ORENCIA intravenously in clinical trials, there were two cases of anaphylaxis or anaphylactoid reactions. Other events potentially associated with drug hypersensitivity, such as hypotension, urticaria, and dyspnea, each occurred in less than 0.9% of ORENCIA-treated patients. Of the 190 patients with juvenile idiopathic arthritis treated with ORENCIA in clinical trials, there was one case of a hypersensitivity reaction (0.5%). Appropriate medical support measures for the treatment of hypersensitivity reactions should be available for immediate use in the event of a reaction [see Adverse Reactions (6.1, 6.3)]. 5.3 Infections Physicians should exercise caution when considering the use of ORENCIA in patients with a history of recurrent infections, underlying conditions which may predispose them to infections, or chronic, latent, or localized infections. Patients who develop a new infection while undergoing treatment with ORENCIA should be monitored closely. Administration of ORENCIA should be discontinued if a patient develops a serious infection [see Adverse Reactions (6.1)]. A higher rate of serious infections has been observed in adult RA patients treated with concurrent TNF antagonists and ORENCIA [see Warnings and Precautions (5.1)]. Prior to initiating immunomodulatory therapies, including ORENCIA, patients should be screened for latent tuberculosis infection with a tuberculin skin test. ORENCIA has not been studied in patients with a positive tuberculosis screen, and the safety of ORENCIA in individuals with latent tuberculosis infection is unknown. Patients testing positive in tuberculosis screening should be treated by standard medical practice prior to therapy with ORENCIA. Antirheumatic therapies have been associated with hepatitis B reactivation. Therefore, screening for viral hepatitis should be performed in accordance with published guidelines before starting therapy with ORENCIA. In clinical studies with ORENCIA, patients who screened positive for hepatitis were excluded from study. 5.4 Immunizations Live vaccines should not be given concurrently with ORENCIA or within 3 months of its discontinuation. No data are available on the secondary transmission of infection from persons receiving live vaccines to patients receiving ORENCIA. The efficacy of vaccination in patients receiving ORENCIA is not known. Based on its mechanism of action, ORENCIA may blunt the effectiveness of some immunizations. It is recommended that patients with juvenile idiopathic arthritis be brought up to date with all immunizations in agreement with current immunization guidelines prior to initiating ORENCIA therapy. 5.5 Use in Patients with Chronic Obstructive Pulmonary Disease (COPD) Adult COPD patients treated with ORENCIA developed adverse events more frequently than those treated with placebo, including COPD exacerbations, cough, rhonchi, and dyspnea. Use of ORENCIA in patients with RA and COPD should be undertaken with caution and such patients should be monitored for worsening of their respiratory status [see Adverse Reactions (6.1)]. 5.6 Immunosuppression The possibility exists for drugs inhibiting T cell activation, including ORENCIA, to affect host defenses against infections and malignancies since T cells mediate cellular immune responses. The impact of treatment with ORENCIA on the development and course of malignancies is not fully understood [see Adverse Reactions (6.1)]. In clinical trials in patients with adult RA, a higher rate of infections was seen in ORENCIA-treated patients compared to placebo [see Adverse Reactions (6.1)]. 6 ADVERSE REACTIONS 6.1 Clinical Studies Experience in Adult RA Patients Treated with Intravenous ORENCIA Because clinical trials are conducted under widely varying and controlled conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not predict the rates observed in a broader patient population in clinical practice. The data described herein reflect exposure to ORENCIA administered intravenously in patients with active RA in placebo-controlled studies (1955 patients with ORENCIA, 989 with placebo). The studies had either a double-blind, placebo-controlled period of 6 months (258 patients with ORENCIA, 133 with placebo) or 1 year (1697 patients with ORENCIA, 856 with placebo). A subset of these patients received concomitant biologic DMARD therapy, such as a TNF blocking agent (204 patients with ORENCIA, 134 with placebo). The majority of patients in RA clinical studies received one or more of the following concomitant medications with ORENCIA: methotrexate, nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, TNF blocking agents, azathioprine, chloroquine, gold, hydroxychloroquine, leflunomide, sulfasalazine, and anakinra. The most serious adverse reactions were serious infections and malignancies. The most commonly reported adverse events (occurring in ≥10% of patients treated with ORENCIA) were headache, upper respiratory tract infection, nasopharyngitis, and nausea. The adverse events most frequently resulting in clinical intervention (interruption or discontinuation of ORENCIA) were due to infection. The most frequently reported infections resulting in dose interruption were upper respiratory tract infection (1.0%), bronchitis (0.7%), and herpes zoster (0.7%). The most frequent infections resulting in discontinuation were pneumonia (0.2%), localized infection (0.2%), and bronchitis (0.1%). Infections In the placebo-controlled trials, infections were reported in 54% of ORENCIA-treated patients and 48% of placebo-treated patients. The most commonly reported infections (reported in 5-13% of patients) were upper respiratory tract infection, nasopharyngitis, sinusitis, urinary tract infection, influenza, and bronchitis. Other infections reported in fewer than 5% of patients at a higher frequency (>0.5%) with ORENCIA compared to placebo, were rhinitis, herpes simplex, and pneumonia [see Warnings and Precautions (5.3)]. Serious infections were reported in 3.0% of patients treated with ORENCIA and 1.9% of patients treated with placebo. The most common (0.2-0.5%) serious infections reported with ORENCIA were pneumonia, cellulitis, urinary tract infection, bronchitis, diverticulitis, and acute pyelonephritis [see Warnings and Precautions (5.3)]. Malignancies In the placebo-controlled portions of the clinical trials (1955 patients treated with ORENCIA for a median of 12 months), the overall frequencies of malignancies were similar in the ORENCIA- and placebo-treated patients (1.3% and 1.1%, respectively). However, more cases of lung cancer were observed in ORENCIA-treated patients (4, 0.2%) than placebo-treated patients (0). In the cumulative ORENCIA clinical trials (placebo-controlled and uncontrolled, open-label) a total of 8 cases of lung cancer (0.21 cases per 100 patient-years) and 4 lymphomas (0.10 cases per 100 patient-years) were observed in 2688 patients (3827 patient-years). The rate observed for lymphoma is approximately 3.5-fold higher than expected in an age- and gender-matched general population based on the National Cancer Institute's Surveillance, Epidemiology, and End Results Database. Patients with RA, particularly those with highly active disease, are at a higher risk for the development of lymphoma. Other malignancies included skin, breast, bile duct, bladder, cervical, endometrial, lymphoma, melanoma, myelodysplastic syndrome, ovarian, prostate, renal, thyroid, and uterine cancers [see Warnings and Precautions(5.6)]. The potential role of ORENCIA in the development of malignancies in humans is unknown. Infusion-Related Reactions and Hypersensitivity Reactions Acute infusion-related events (adverse reactions occurring within 1 hour of the start of the infusion) in Studies III, IV, and V [see Clinical Studies (14.1)] were more common in the ORENCIA-treated patients than the placebo patients (9% for ORENCIA, 6% for placebo). The most frequently reported events (1-2%) were dizziness, headache, and hypertension. Acute infusion-related events that were reported in >0.1% and ≤1% of patients treated with ORENCIA included cardiopulmonary symptoms, such as hypotension, increased blood pressure, and dyspnea; other symptoms included nausea, flushing, urticaria, cough, hypersensitivity, pruritus, rash, and wheezing. Most of these reactions were mild (68%) to moderate (28%). Fewer than 1% of ORENCIA-treated patients discontinued due to an acute infusion-related event. In controlled trials, 6 ORENCIA-treated patients compared to 2 placebo-treated patients discontinued study treatment due to acute infusion-related events. Of 2688 patients treated with ORENCIA in clinical trials, there were two cases of anaphylaxis or anaphylactoid reactions. Other events potentially associated with drug hypersensitivity, such as hypotension, urticaria, and dyspnea, each occurred in less than 0.9% of ORENCIA-treated patients and generally occurred within 24 hours of ORENCIA infusion. Appropriate medical support measures for the treatment of hypersensitivity reactions should be available for immediate use in the event of a reaction [see Warnings and Precautions (5.2)]. Adverse Reactions in Patients with COPD In Study V [see Clinical Studies (14.1)], there were 37 patients with chronic obstructive pulmonary disease (COPD) who were treated with ORENCIA and 17 COPD patients who were treated with placebo. The COPD patients treated with ORENCIA developed adverse events more frequently than those treated with placebo (97% vs 88%, respectively). Respiratory disorders occurred more frequently in ORENCIA-treated patients compared to placebo-treated patients (43% vs 24%, respectively) including COPD exacerbation, cough, rhonchi, and dyspnea. A greater percentage of ORENCIA-treated patients developed a serious adverse event compared to placebo-treated patients (27% vs 6%), including COPD exacerbation (3 of 37 patients [8%]) and pneumonia (1 of 37 patients [3%]) [see Warnings and Precautions (5.5)]. Other Adverse Reactions Adverse events occurring in 3% or more of patients and at least 1% more frequently in ORENCIA-treated patients during placebo-controlled RA studies are summarized in Table 2.
Antibodies directed against the entire abatacept molecule or to the CTLA-4 portion of abatacept were assessed by ELISA assays in RA patients for up to 2 years following repeated treatment with ORENCIA. Thirty-four of 1993 (1.7%) patients developed binding antibodies to the entire abatacept molecule or to the CTLA-4 portion of abatacept. Because trough levels of abatacept can interfere with assay results, a subset analysis was performed. In this analysis it was observed that 9 of 154 (5.8%) patients that had discontinued treatment with ORENCIA for over 56 days developed antibodies. Samples with confirmed binding activity to CTLA-4 were assessed for the presence of neutralizing antibodies in a cell-based luciferase reporter assay. Six of 9 (67%) eva luable patients were shown to possess neutralizing antibodies. However, the development of neutralizing antibodies may be underreported due to lack of assay sensitivity. No correlation of antibody development to clinical response or adverse events was observed. The data reflect the percentage of patients whose test results were positive for antibodies to abatacept in specific assays. The observed incidence of antibody (including neutralizing antibody) positivity in an assay is highly dependent on several factors, including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medication, and underlying disease. For these reasons, comparison of the incidence of antibodies to abatacept with the incidence of antibodies to other products may be misleading. Clinical Experience in Methotrexate-Naive Patients Study VI was an active-controlled clinical trial in methotrexate-naive patients [see Clinical Studies (14.1)]. The safety experience in these patients was consistent with Studies I-V. 6.2 Clinical Experience in Adult RA Patients Treated with Subcutaneous ORENCIA Because clinical trials are conducted under widely varying and controlled conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not predict the rates observed in a broader patient population in clinical practice. Study SC-I was a randomized, double-blind, double-dummy, non-inferiority study that compared the efficacy and safety of abatacept administered subcutaneously (SC) and intravenously (IV) in 1457 subjects with rheumatoid arthritis, receiving background methotrexate, and experiencing an inadequate response to methotrexate (MTX-IR) [see Clinical Studies (14.1)]. The safety experience and immunogenicity for ORENCIA administered subcutaneously was consistent with intravenous Studies I-VI. Due to the route of administration, injection site reactions and immunogenicity were eva luated in Study SC-I and two other smaller studies discussed in the sections below. Injection Site Reactions in Adult RA Patients Treated with Subcutaneous ORENCIA Study SC-I compared the safety of abatacept including injection site reactions following subcutaneous or intravenous administration. The overall frequency of injection site reactions was 2.6% (19/736) and 2.5% (18/721) for the subcutaneous abatacept group and the intravenous abatacept group (subcutaneous placebo), respectively. All these injection site reactions (including hematoma, pruritus, and erythema) were mild (83%) to moderate (17%) in severity, and none necessitated drug discontinuation. Immunogenicity in Adult RA Patients Treated with Subcutaneous ORENCIA Study SC-I compared the immunogenicity to abatacept following subcutaneous or intravenous administration. The overall immunogenicity frequency to abatacept was 1.1% (8/725) and 2.3% (16/710) for the subcutaneous and intravenous groups, respectively. The rate is consistent with previous experience, and there was no correlation of immunogenicity with effects on pharmacokinetics, safety, or efficacy. Immunogenicity and Safety of Subcutaneous ORENCIA Administration as Monotherapy without an Intravenous Loading Dose Study SC-II was conducted to determine the effect of monotherapy use of ORENCIA on immunogenicity following subcutaneous administration without an intravenous load in 100 RA patients, who had not previously received abatacept or other CTLA4Ig, who received either subcutaneous ORENCIA plus methotrexate (n=51) or subcutaneous ORENCIA monotherapy (n=49). No patients in either group developed anti-product antibodies after 4 months of treatment. The safety observed in this study was consistent with that observed in the other subcutaneous studies. Immunogenicity and Safety of Subcutaneous ORENCIA upon Withdrawal (Three Months) and Restart of Treatment Study SC-III in the subcutaneous program was conducted to investigate the effect of withdrawal (three months) and restart of ORENCIA subcutaneous treatment on immunogenicity in RA patients treated concomitantly with methotrexate. One hundred sixty-seven patients were enrolled in the first 3-month treatment period and responders (n=120) were randomized to either subcutaneous ORENCIA or placebo for the second 3-month period (withdrawal period). Patients from this period then received open-label ORENCIA treatment in the final 3-month period of the study (period 3). At the end of the withdrawal period, 0/38 patients who continued to receive subcutaneous ORENCIA developed anti-product antibodies compared to 7/73 (9.6%) of patients who had subcutaneous ORENCIA withdrawn during this period. Half of the patients receiving subcutaneous placebo during the withdrawal period received a single intravenous infusion of ORENCIA at the start of period 3 and half received intravenous placebo. At the end of period 3, when all patients again received subcutaneous ORENCIA, the immunogenicity rates were 1/38 (2.6%) in the group receiving subcutaneous ORENCIA throughout, and 2/73 (2.7%) in the group that had received placebo during the withdrawal period. Upon reinitiating therapy, there were no injection reactions and no differences in response to therapy in patients who were withdrawn from subcutaneous therapy for up to 3 months relative to those who remained on subcutaneous therapy, whether therapy was reintroduced with or without an intravenous loading dose. The safety observed in this study was consistent with that observed in the other studies. 6.3 Clinical Studies Experience in Juvenile Idiopathic Arthritis In general, the adverse events in pediatric patients were similar in frequency and type to those seen in adult patients [see Warnings and Precautions (5), Adverse Reactions (6)]. ORENCIA has been studied in 190 pediatric patients, 6 to 17 years of age, with polyarticular juvenile idiopathic arthritis. Overall frequency of adverse events in the 4-month, lead-in, open-label period of the study was 70%; infections occurred at a frequency of 36% [see Clinical Studies (14.2)]. The most common infections were upper respiratory tract infection and nasopharyngitis. The infections resolved without sequelae, and the types of infections were consistent with those commonly seen in outpatient pediatric populations. Other events that occurred at a preva lence of at least 5% were headache, nausea, diarrhea, cough, pyrexia, and abdominal pain. A total of 6 serious adverse events (acute lymphocytic leukemia, ovarian cyst, varicella infection, disease flare [2], and joint wear) were reported during the initial 4 months of treatment with ORENCIA. Of the 190 patients with juvenile idiopathic arthritis treated with ORENCIA in clinical trials, there was one case of a hypersensitivity reaction (0.5%). During Periods A, B, and C, acute infusion-related reactions occurred at a frequency of 4%, 2%, and 3%, respectively, and were consistent with the types of events reported in adults. Upon continued treatment in the open-label extension period, the types of adverse events were similar in frequency and type to those seen in adult patients, except for a single patient diagnosed with multiple sclerosis while on open-label treatment. Immunogenicity Antibodies directed against the entire abatacept molecule or to the CTLA-4 portion of abatacept were assessed by ELISA assays in patients with juvenile idiopathic arthritis following repeated treatment with ORENCIA throughout the open-label period. For patients who were withdrawn from therapy for up to 6 months during the double-blind period, the rate of antibody formation to the CTLA-4 portion of the molecule was 41% (22/54), while for those who remained on therapy the rate was 13% (7/54). Twenty of these patients had samples that could be tested for antibodies with neutralizing activity; of these, 8 (40%) patients were shown to possess neutralizing antibodies. The presence of antibodies was generally transient and titers were low. The presence of antibodies was not associated with adverse events, changes in efficacy, or an effect on serum concentrations of abatacept. For patients who were withdrawn from ORENCIA during the double-blind period for up to 6 months, no serious acute infusion-related events were observed upon re-initiation of ORENCIA therapy. 6.4 Postmarketing Experience Adverse reactions have been reported during the postapproval use of ORENCIA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to ORENCIA. Based on the postmarketing experience in adult RA patients, the following adverse reaction has been identified during postapproval use with ORENCIA.
7 DRUG INTERACTIONS 7.1 TNF Antagonists Concurrent administration of a TNF antagonist with ORENCIA has been associated with an increased risk of serious infections and no significant additional efficacy over use of the TNF antagonists alone. Concurrent therapy with ORENCIA and TNF antagonists is not recommended [see Warnings and Precautions (5.1)]. 7.2 Other Biologic RA Therapy There is insufficient experience to assess the safety and efficacy of ORENCIA administered concurrently with other biologic RA therapy, such as anakinra, and therefore such use is not recommended. 7.3 Blood Glucose Testing Parenteral drug products containing maltose can interfere with the readings of blood glucose monitors that use test strips with glucose dehydrogenase pyrroloquinolinequinone (GDH-PQQ). The GDH-PQQ based glucose monitoring systems may react with the maltose present in ORENCIA for intravenous administration, resulting in falsely elevated blood glucose readings on the day of infusion. When receiving ORENCIA through intravenous administration, patients that require blood glucose monitoring should be advised to consider methods that do not react with maltose, such as those based on glucose dehydrogenase nicotine adenine dinucleotide (GDH-NAD), glucose oxidase, or glucose hexokinase test methods. ORENCIA for subcutaneous administration does not contain maltose; therefore, patients do not need to alter their glucose monitoring. 8 USE IN SPECIFIC POPULATIONS 8.1 Pregnancy Pregnancy Category C There are no adequate and well-controlled studies of ORENCIA use in pregnant women. Abatacept has been shown to cross the placenta in animals, and in animal reproduction studies alterations in immune function occurred. ORENCIA should be used during pregnancy only if the potential benefit to the mother justifies the potential risk to the fetus. Abatacept was not teratogenic when administered to pregnant mice at doses up to 300 mg/kg and in pregnant rats and rabbits at doses up to 200 mg/kg daily representing approximately 29 times the exposure associated with the maximum recommended human dose (MRHD) of 10 mg/kg based on AUC (area under the time-concentration curve). Abatacept administered to female rats every three days during early gestation and throughout the lactation period, produced no adverse effects in offspring at doses up to 45 mg/kg, representing 3 times the exposure associated with the MRHD of 10 mg/kg based on AUC. However, at 200 mg/kg, 11 times the MRHD exposure, alterations in immune function were observed consisting of a 9-fold increase in T-cell dependent antibody response in female pups and thyroid inflammation in one female pup. It is not known whether these findings indicate a risk for development of autoimmune diseases in humans exposed in utero to abatacept. However, exposure to abatacept in the juvenile rat, which may be more representative of the fetal immune system state in the human, resulted in immune system abnormalities including inflammation of the thyroid and pancreas [see Nonclinical Toxicology (13.2)]. Pregnancy Registry: To monitor maternal-fetal outcomes of pregnant women exposed to ORENCIA, a pregnancy registry has been established. Healthcare professionals are encouraged to register patients and pregnant women are encouraged to enroll themselves by calling 1-877-311-8972. 8.3 Nursing Mothers It is not known whether ORENCIA is excreted into human milk or absorbed systemically after ingestion by a nursing infant. However, abatacept was excreted in rat milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from ORENCIA, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. 8.4 Pediatric Use ORENCIA is indicated for reducing signs and symptoms in pediatric patients with moderately to severely active polyarticular juvenile idiopathic arthritis ages 6 years and older. ORENCIA may be used as monotherapy or concomitantly with methotrexate. Studies in juvenile rats exposed to ORENCIA prior to immune system maturity have shown immune system abnormalities including an increase in the incidence of infections leading to death as well as inflammation of the thyroid and pancreas [see Nonclinical Toxicology (13.2)]. Studies in adult mice and monkeys have not demonstrated similar findings. As the immune system of the rat is undeveloped in the first few weeks after birth, the relevance of these results to humans greater than 6 years of age (where the immune system is largely developed) is unknown. ORENCIA is not recommended for use in patients below the age of 6 years. The safety and effectiveness of ORENCIA in pediatric patients below 6 years of age have not been established. The safety and efficacy of ORENCIA in pediatric patients for uses other than juvenile idiopathic arthritis have not been established. 8.5 Geriatric Use A total of 323 patients 65 years of age and older, including 53 patients 75 years and older, received ORENCIA in clinical studies. No overall differences in safety or effectiveness were observed between these patients and younger patients, but these numbers are too low to rule out differences. The frequency of serious infection and malignancy among ORENCIA-treated patients over age 65 was higher than for those under age 65. Because there is a higher incidence of infections and malignancies in the elderly population in general, caution should be used when treating the elderly. 10 OVERDOSAGE Doses up to 50 mg/kg have been administered intravenously without apparent toxic effect. In case of overdosage, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions and appropriate symptomatic treatment instituted. 11 DESCRIPTION ORENCIA® (abatacept) is a soluble fusion protein that consists of the extracellular domain of human cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) linked to the modified Fc (hinge, CH2, and CH3 domains) portion of human immunoglobulin G1 (IgG1). Abatacept is produced by recombinant DNA technology in a mammalian cell expression system. The apparent molecular weight of abatacept is 92 kilodaltons. ORENCIA lyophilized powder for intravenous infusion is supplied as a sterile, white, preservative-free, lyophilized powder for intravenous administration. Following reconstitution of the lyophilized powder with 10 mL of Sterile Water for Injection, USP, the solution of ORENCIA is clear, colorless to pale yellow, with a pH range of 7.2 to 7.8. Each single-use vial of ORENCIA provides 250 mg abatacept, maltose (500 mg), monobasic sodium phosphate (17.2 mg), and sodium chloride (14.6 mg) for administration. ORENCIA solution for subcutaneous administration is supplied as a sterile, preservative-free, clear, colorless to pale yellow solution with a pH of 6.8 to 7.4. Each single dose of subcutaneous injection provides 125 mg abatacept, dibasic sodium phosphate anhydrous (0.838 mg), monobasic sodium phosphate monohydrate (0.286 mg), poloxamer 188 (8 mg), sucrose (170 mg), and quantity sufficient to 1 mL with water for injection. Unlike the intravenous formulation, ORENCIA solution for subcutaneous administration contains no maltose. 12 CLINICAL PHARMACOLOGY 12.1 Mechanism of Action Abatacept, a selective costimulation modulator, inhibits T cell (T lymphocyte) activation by binding to CD80 and CD86, thereby blocking interaction with CD28. This interaction provides a costimulatory signal necessary for full activation of T lymphocytes. Activated T lymphocytes are implicated in the pathogenesis of RA and are found in the synovium of patients with RA. In vitro, abatacept decreases T cell proliferation and inhibits the production of the cytokines TNF alpha (TNFα), interferon-γ, and interleukin-2. In a rat collagen-induced arthritis model, abatacept suppresses inflammation, decreases anti-collagen antibody production, and reduces antigen specific production of interferon-γ. The relationship of these biological response markers to the mechanisms by which ORENCIA exerts its effects in RA is unknown. 12.2 Pharmacodynamics In clinical trials with ORENCIA at doses approximating 10 mg/kg, decreases were observed in serum levels of soluble interleukin-2 receptor (sIL-2R), interleukin-6 (IL-6), rheumatoid factor (RF), C-reactive protein (CRP), matrix metalloproteinase-3 (MMP3), and TNFα. The relationship of these biological response markers to the mechanisms by which ORENCIA exerts its effects in RA is unknown. 12.3 Pharmacokinetics Healthy Adults and Adult RA - Intravenous Administration The pharmacokinetics of abatacept were studied in healthy adult subjects after a single 10 mg/kg intravenous infusion and in RA patients after multiple 10 mg/kg intravenous infusions (see Table 3).
The pharmacokinetics of abatacept in RA patients and healthy subjects appeared to be comparable. In RA patients, after multiple intravenous infusions, the pharmacokinetics of abatacept showed proportional increases of Cmax and AUC over the dose range of 2 mg/kg to 10 mg/kg. At 10 mg/kg, serum concentration appeared to reach a steady-state by day 60 with a mean (range) trough concentration of 24 (1 to 66) mcg/mL. No systemic accumulation of abatacept occurred upon continued repeated treatment with 10 mg/kg at monthly intervals in RA patients. Population pharmacokinetic analyses in RA patients revealed that there was a trend toward higher clearance of abatacept with increasing body weight. Age and gender (when corrected for body weight) did not affect clearance. Concomitant methotrexate, NSAIDs, corticosteroids, and TNF blocking agents did not influence abatacept clearance. No formal studies were conducted to examine the effects of either renal or hepatic impairment on the pharmacokinetics of abatacept. Juvenile Idiopathic Arthritis In patients 6 to 17 years of age, the mean (range) steady-state serum peak and trough concentrations of abatacept were 217 (57 to 700) and 11.9 (0.15 to 44.6) mcg/mL. Population pharmacokinetic analyses of the serum concentration data showed that clearance of abatacept increased with baseline body weight. The estimated mean (range) clearance of abatacept in the juvenile idiopathic arthritis patients was 0.4 (0.20 to 1.12) mL/h/kg. After accounting for the effect of body weight, the clearance of abatacept was not related to age and gender. Concomitant methotrexate, corticosteroids, and NSAIDs were also shown not to influence abatacept clearance. Adult RA - Subcutaneous Administration Abatacept exhibited linear pharmacokinetics following subcutaneous administration. The mean (range) for Cmin and Cmax at steady state observed after 85 days of treatment was 32.5 mcg/mL (6.6 to 113.8 mcg/mL) and 48.1 mcg/mL (9.8 to 132.4 mcg/mL), respectively. The bioavailability of abatacept following subcutaneous administration relative to intravenous administration is 78.6%. Mean estimates for systemic clearance (0.28 mL/h/kg), volume of distribution (0.11 L/kg), and terminal half-life (14.3 days) were comparable between subcutaneous and intravenous administration. Study SC-II was conducted to determine the effect of monotherapy use of ORENCIA on immunogenicity following subcutaneous administration without an intravenous load. When the intravenous loading dose was not administered, a mean trough concentration of 12.6 mcg/mL was achieved after 2 weeks of dosing. Consistent with the intravenous data, population pharmacokinetic analyses for subcutaneous abatacept in RA patients revealed that there was a trend toward higher clearance of abatacept with increasing body weight. Age and gender (when corrected for body weight) did not affect apparent clearance. Concomitant medication, such as methotrexate, corticosteroids, and NSAIDs, did not influence abatacept apparent clearance. 13 NONCLINICAL TOXICOLOGY 13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility In a mouse carcinogenicity study, weekly subcutaneous injections of 20, 65, or 200 mg/kg of abatacept administered for up to 84 weeks in males and 88 weeks in females were associated with increases in the incidence of malignant lymphomas (all doses) and mammary gland tumors (intermediate- and high-dose in females). The mice from this study were infected with murine leukemia virus and mouse mammary tumor virus. These viruses are associated with an increased incidence of lymphomas and mammary gland tumors, respectively, in immunosuppressed mice. The doses used in these studies produced exposures 0.8, 2.0, and 3.0 times higher, respectively, than the exposure associated with the maximum recommended human dose (MRHD) of 10 mg/kg based on AUC (area under the time-concentration curve). The relevance of these findings to the clinical use of ORENCIA is unknown. In a one-year toxicity study in cynomolgus monkeys, abatacept was administered intravenously once weekly at doses up to 50 mg/kg (producing 9 times the MRHD exposure based on AUC). Abatacept was not associated with any significant drug-related toxicity. Reversible pharmacological effects consisted of minimal transient decreases in serum IgG and minimal to severe lymphoid depletion of germinal centers in the spleen and/or lymph nodes. No evidence of lymphomas or preneoplastic morphologic changes was observed, despite the presence of a virus (lymphocryptovirus) known to cause these lesions in immunosuppressed monkeys within the time frame of this study. The relevance of these findings to the clinical use of ORENCIA is unknown. No mutagenic potential of abatacept was observed in the in vitro bacterial reverse mutation (Ames) or Chinese hamster ovary/hypoxanthine guanine phosphoribosyl-transferase (CHO/HGPRT) forward point mutation assays with or without metabolic activation, and no chromosomal aberrations were observed in human lymphocytes treated with abatacept with or without metabolic activation. Abatacept had no adverse effects on male or female fertility in rats at doses up to 200 mg/kg every three days (11 times the MRHD exposure based on AUC). 13.2 Animal Toxicology and/or Pharmacology A juvenile animal study was conducted in rats dosed with abatacept from 4 to 94 days of age in which an increase in the incidence of infections leading to death occurred at all doses compared with controls. Altered T-cell subsets including increased T-helper cells and reduced T-regulatory cells were observed. In addition, inhibition of T-cell-dependent antibody responses (TDAR) was observed. Upon following these animals into adulthood, lymphocytic inflammation of the thyroid and pancreatic islets wasobserved. In studies of adult mice and monkeys, inhibition of TDAR was apparent. However, infection and mortality, altered T-helper cells, and inflammation of thyroid and pancreas were not observed. 14 CLINICAL STUDIES 14.1 Adult Rheumatoid Arthritis The efficacy and safety of ORENCIA for intravenous administration were assessed in six randomized, double-blind, controlled studies (five placebo-controlled and one active-controlled) in patients ≥18 years of age with active RA diagnosed according to American College of Rheumatology (ACR) criteria. Studies I, II, III, IV, and VI required patients to have at least 12 tender and 10 swollen joints at randomization. Study V did not require any specific number of tender or swollen joints. ORENCIA or placebo treatment was given intravenously at weeks 0, 2, and 4 and then every 4 weeks thereafter in intravenous Studies I, II, III, IV, and VI. The safety and efficacy of ORENCIA for subcutaneous administration were assessed in Study SC-I, which was a randomized, double-blind, double-dummy, non-inferiority study that compared abatacept administered subcutaneously and intravenously in 1457 subjects with rheumatoid arthritis (RA), receiving background methotrexate (MTX), and experiencing an inadequate response to methotrexate (MTX-IR). Study I eva luated ORENCIA as monotherapy in 122 patients with active RA who had failed at least one non-biologic DMARD or etanercept. In Study II and Study III, the efficacy and safety of ORENCIA were assessed in patients with an inadequate response to methotrexate and who were continued on their stable dose of methotrexate. In Study IV, the efficacy and safety of ORENCIA were assessed in patients with an inadequate response to a TNF blocking agent, with the TNF blocking agent discontinued prior to randomization; other DMARDs were permitted. Study V primarily assessed safety in patients with active RA requiring additional intervention in spite of current therapy with DMARDs; all DMARDs used at enrollment were continued. Patients in Study V were not excluded for comorbid medical conditions. In Study VI, the efficacy and safety of ORENCIA were assessed in methotrexate-naive patients with RA of less than 2 years disease duration. In Study VI, patients previously naive to methotrexate were randomized to receive ORENCIA plus methotrexate or methotrexate plus placebo. In Study SC-I, the goal was to demonstrate the efficacy and safety of ORENCIA subcutaneous relative to ORENCIA intravenous administration in subjects with moderate to severely active RA and experiencing inadequate response to methotrexate, using a non-inferiority study design. Study I patients were randomized to receive one of three doses of ORENCIA (0.5, 2, or 10 mg/kg) or placebo ending at week 8. Study II patients were randomized to receive ORENCIA 2 or 10 mg/kg or placebo for 12 months. Study III, IV, V, and VI patients were randomized to receive a dose of ORENCIA based on weight range or placebo for 12 months (Studies III, V, and VI) or 6 months (Study IV). The dose of ORENCIA was 500 mg for patients weighing less than 60 kg, 750 mg for patients weighing 60 to 100 kg, and 1000 mg for patients weighing greater than 100 kg. In Study SC-I, patients were randomized with stratification by body weight (<60 kg, 60 to 100 kg, >100 kg) to receive ORENCIA 125 mg subcutaneous injections weekly, after a single intravenous loading dose of ORENCIA based on body weight or ORENCIA intravenously on Days 1, 15, 29, and every four weeks thereafter. Subjects continued taking their current dose of methotrexate from the day of randomization. Clinical Response The percent of ORENCIA-treated patients achieving ACR 20, 50, and 70 responses and major clinical response in Studies I, III, IV, and VI are shown in Table 4. ORENCIA-treated patients had higher ACR 20, 50, and 70 response rates at 6 months compared to placebo-treated patients. Month 6 ACR response rates in Study II for the 10 mg/kg group were similar to the ORENCIA group in Study III. In Studies III and IV, improvement in the ACR 20 response rate versus placebo was observed within 15 days in some patients and within 29 days versus methotrexate in Study VI. In Studies II, III, and VI, ACR response rates were maintained to 12 months in ORENCIA-treated patients. ACR responses were maintained up to three years in the open-label extension of Study II. In Study III, ORENCIA-treated patients experienced greater improvement than placebo-treated patients in morning stiffness. In Study VI, a greater proportion of patients treated with ORENCIA plus methotrexate achieved a low level of disease activity as measured by a DAS28-CRP less than 2.6 at 12 months compared to those treated with methotrexate plus placebo (Table 4). Of patients treated with ORENCIA plus methotrexate who achieved DAS28-CRP less than 2.6, 54% had no active joints, 17% had one active joint, 7% had two active joints, and 22% had three or more active joints, where an active joint was a joint that was rated as tender or swollen or both. In Study SC-I, the main outcome measure was ACR 20 at 6 months. The pre-specified non-inferiority margin was a treatment difference of −7.5%. As shown in Table 4, the study demonstrated non-inferiority of ORENCIA administered subcutaneously to intravenous infusions of ORENCIA with respect to ACR 20 responses up to 6 months of treatment. ACR 50 and 70 responses are also shown in Table 4. No major differences in ACR responses were observed between intravenous and subcutaneous treatment groups in subgroups based on weight categories (less than 60 kg, 60 to 100 kg, and more than 100 kg; data not shown).
The results of the components of the ACR response criteria for Studies III, IV, and SC-I are shown in Table 5 (results at Baseline [BL] and 6 months [6 M]). In ORENCIA-treated patients, greater improvement was seen in all ACR response criteria components through 6 and 12 months than in placebo-treated patients.
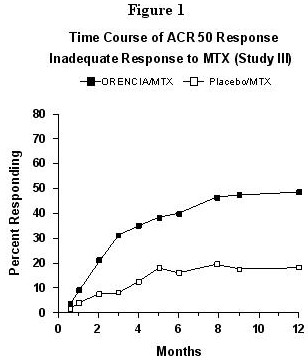
The percent of patients achieving the ACR 50 response for Study III by visit is shown in Figure1. The time course for the ORENCIA group in Study VI was similar to that in Study III. Figure 1: Percent of Patients Achieving ACR 50 Response by Visit* (Study III)
*The same patients may not have responded at each time point.
The percent of patients achieving the ACR 50 response for Study SC-I in the ORENCIA subcutaneous (SC) and intravenous (IV) treatment arms at each treatment visit was as follows: Day 15—SC 3%, IV 5%; Day 29—SC 11%, IV 14%; Day 57—SC 24%, IV 30%; Day 85—SC 33%, IV 38%; Day 113—SC 39%, IV 41%; Day 141—SC 46%, IV 47%; Day 169—SC 51%, IV 50%. Radiographic Response In Study III and Study VI, structural joint damage was assessed radiographically and expressed as change from baseline in the Genant-modified Total Sharp Score (TSS) and its components, the Erosion Score (ES) and Joint Space Narrowing (JSN) score. ORENCIA/methotrexate slowed the progression of structural damage compared to placebo/methotrexate after 12 months of treatment as shown in Table 6.
In the open-label extension of Study III, 75% of patients initially randomized to ORENCIA/methotrexate and 65% of patients initially randomized to placebo/methotrexate were eva luated radiographically at Year 2. As shown in Table 6, progression of structural damage in ORENCIA/methotrexate-treated patients was further reduced in the second year of treatment. Following 2 years of treatment with ORENCIA/methotrexate, 51% of patients had no progression of structural damage as defined by a change in the TSS of zero or less compared with baseline. Fifty-six percent (56%) of ORENCIA/methotrexate-treated patients had no progression during the first year compared to 45% of placebo/methotrexate-treated patients. In their second year of treatment with ORENCIA/methotrexate, more patients had no progression than in the first year (65% vs 56%). Physical Function Response and Health-Related Outcomes Improvement in physical function was measured by the Health Assessment Questionnaire Disability Index (HAQ-DI). In the HAQ-DI, ORENCIA demonstrated greater improvement from baseline versus placebo in Studies II-V and versus methotrexate in Study VI. In Study SC-I, improvement from baseline as measured by HAQ-DI at 6 months and over time was similar between subcutaneous and intravenous administration. The results from Studies II and III are shown in Table 7. Similar results were observed in Study V compared to placebo and in Study VI compared to methotrexate. During the open-label period of Study II, the improvement in physical function has been maintained for up to 3 years.
Health-related quality of life was assessed by the SF-36 questionnaire at 6 months in Studies II, III, and IV and at 12 months in Studies II and III. In these studies, improvement was observed in the ORENCIA group as compared with the placebo group in all 8 domains of the SF-36 as well as the Physical Component Summary (PCS) and the Mental Component Summary (MCS). 14.2 Juvenile Idiopathic Arthritis The safety and efficacy of ORENCIA were assessed in a three-part study including an open-label extension in children with polyarticular juvenile idiopathic arthritis (JIA). Patients 6 to 17 years of age (n=190) with moderately to severely active polyarticular JIA who had an inadequate response to one or more DMARDs, such as methotrexate or TNF antagonists, were treated. Patients had a disease duration of approximately 4 years with moderately to severely active disease at study entry, as determined by baseline counts of active joints (mean, 16) and joints with loss of motion (mean, 16); patients had elevated C-reactive protein (CRP) levels (mean, 3.2 mg/dL) and ESR (mean, 32 mm/h). The patients enrolled had subtypes of JIA that at disease onset included Oligoarticular (16%), Polyarticular (64%; 20% were rheumatoid factor positive), and Systemic (20%). At study entry, 74% of patients were receiving methotrexate (mean dose, 13.2 mg/m2 per week) and remained on a stable dose of methotrexate (those not receiving methotrexate did not initiate methotrexate treatment during the study). In Period A (open-label, lead-in), patients received 10 mg/kg (maximum 1000 mg per dose) intravenously on days 1, 15, 29, and monthly thereafter. Response was assessed utilizing the ACR Pediatric 30 definition of improvement, defined as ≥30% improvement in at least 3 of the 6 JIA core set variables and ≥30% worsening in not more than 1 of the 6 JIA core set variables. Patients demonstrating an ACR Pedi 30 response at the end of Period A were randomized into the double-blind phase (Period B) and received either ORENCIA or placebo for 6 months or until disease flare. Disease flare was defined as a ≥30% worsening in at least 3 of the 6 JIA core set variables with ≥30% improvement in not more than 1 of the 6 JIA core set variables; ≥2 cm of worsening of the Physician or Parent Global Assessment was necessary if used as 1 of the 3 JIA core set variables used to define flare, and worsening in ≥2 joints was necessary if the number of active joints or joints with limitation of motion was used as 1 of the 3 JIA core set variables used to define flare. At the conclusion of Period A, pediatric ACR 30/50/70 responses were 65%, 50%, and 28%, respectively. Pediatric ACR 30 responses were similar in all subtypes of JIA studied. During the double-blind randomized withdrawal phase (Period B), ORENCIA-treated patients experienced significantly fewer disease flares compared to placebo-treated patients (20% vs 53%); 95% CI of the difference (15%, 52%). The risk of disease flare among patients continuing on ORENCIA was less than one-third than that for patients withdrawn from ORENCIA treatment (hazard ratio=0.31, 95% CI [0.16, 0.59]). Among patients who received ORENCIA throughout the study (Period A, Period B, and the open-label extension Period C), the proportion of pediatric ACR 30/50/70 responders has remained consistent for 1 year. 16 HOW SUPPLIED/STORAGE AND HANDLING For Intravenous Infusion ORENCIA® (abatacept) lyophilized powder for intravenous infusion is supplied as an individually packaged, single-use vial with a silicone-free disposable syringe, providing 250 mg abatacept in a 15-mL vial: NDC 0003-2187-10. For Subcutaneous Injection ORENCIA® (abatacept) injection solution for subcutaneous administration is supplied as a single-dose disposable prefilled glass syringe with flange extender. The Type I glass syringe has a coated stopper and fixed stainless steel needle (5 bevel, 29-gauge thin wall, ½-inch needle) covered with a rigid needle shield. The prefilled syringe provides 125 mg of abatacept in 1 mL and is provided in a pack of 4 syringes: NDC 0003-2188-31. Storage ORENCIA lyophilized powder supplied in a vial should be refrigerated at 2°C to 8°C (36°F to 46°F). Do not use beyond the expiration date on the vial. Protect the vials from light by storing in the original package until time of use. ORENCIA solution supplied in a prefilled syringe should be refrigerated at 2°C to 8°C (36°F to 46°F). Do not use beyond the expiration date on the prefilled syringe. Protect from light by storing in the original package until time of use. Do not allow the prefilled syringe to freeze. 百时美施贵宝阿巴西普(ORENCIA)皮下注射配方获欧盟委员会批准 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||