|
英文药名:Dyloject(diclofenac sodium Injection)
中文药名:双氯芬酸钠注射剂
生产厂家:Hospira, Inc.
药品介绍
2014年12月23日,获FDA批准上市,商品名Dyloject,为双氯芬酸钠的注射剂,规格为37.5毫克/毫升。该药可以单独使用也可以与阿片类药物合用,治疗轻、中、重度的疼痛,每天只需用1次。双氯酸酸钠之前在美国上市的剂型只有普通片剂、缓释片、外用凝胶、滴眼剂以及局部用溶液剂,这是第一次注射剂剂型获批。
双氯芬酸钠(Diclofenac Sodium) 该药由Javelin制药公司申报
本品美国上市:37.5mg/1mL*25瓶/盒:产地:Hospira公司
本品英国上市:75mg/2ml 产地:Therabel制药英国
www.oneyao.net
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use Dyloject safely and effectively. See full prescribing information for Dyloject.
Dyloject™ (diclofenac sodium) Injection, for intravenous use
Initial U.S. Approval: 1988
WARNING: RISK OF SERIOUS CARDIOVASCULAR AND GASTROINTESTINAL EVENTSSee full prescribing information for complete boxed warning.
Cardiovascular Risk
• Non-steroidal anti-inflammatory drugs (NSAIDs) may increase the risk of serious cardiovascular (CV) thrombotic events, myocardial infarction, and stroke, which can be fatal. Risk may increase with duration of use. Patients with cardiovascular disease or risk factors for cardiovascular disease may be at greater risk. (5.1)
• Dyloject is contraindicated for the treatment of perioperative pain in the setting of coronary artery bypass graft (CABG) surgery. (4)
Gastrointestinal Risk
• NSAIDs increase the risk of serious gastrointestinal (GI) adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which can be fatal. Events can occur at any time without warning symptoms. Elderly patients are at greater risk. (5.2)
INDICATIONS AND USAGE
Dyloject is an NSAID indicated for use in adults for the:
• management of mild to moderate pain. ( 1)
• management of moderate to severe pain alone or in combination with opioid analgesics. ( 1)
DOSAGE AND ADMINISTRATION
• 37.5 mg administered by intravenous bolus injection over 15 seconds. Treatment may be repeated every 6 hours, not to exceed 150 mg/day. ( 2)
• Patients must be well hydrated before Dyloject administration. ( 2)
DOSAGE FORMS AND STRENGTHS
Injection, single use vial containing 37.5 mg/mL. (3)
CONTRAINDICATIONS
• Known hypersensitivity to diclofenac. ( 4)
• History of asthma, urticaria, or allergic-type reactions after taking aspirin or other NSAIDs. ( 4)
• Perioperative pain in the setting of coronary artery bypass graft (CABG) surgery. ( 4)
• Moderate to severe renal insufficiency in the perioperative period and who are at risk for volume depletion. ( 4)
WARNINGS AND PRECAUTIONS
• Serious and potentially fatal cardiovascular (CV) thrombotic events, myocardial infarction, and stroke: Patients with known CV disease or risk factors for CV disease may be at greater risk. Use for the shortest possible duration. ( 5.1)
• Serious gastrointestinal (GI) adverse events including bleeding, ulceration, and perforation, which can be fatal: Use for the shortest possible duration. Use with caution in patients with prior history of ulcer disease or GI bleeding. ( 5.2)
• Renal papillary necrosis and other renal injury with long-term administration of NSAIDs: Use Dyloject with caution in patients at greatest risk for this reaction, including the elderly; those with impaired renal function, heart failure, or liver impairment; and those taking diuretics or ACE inhibitors. ( 5.3)
• Elevation of one or more liver tests and severe hepatic reactions: Discontinue Dyloject immediately if abnormal liver tests persist or worsen. ( 5.4)
• New onset or worsening of hypertension: Monitor blood pressure closely during treatment with Dyloject. ( 5.5)
• Fluid retention and edema: Use Dyloject with caution in patients with fluid retention or heart failure. ( 5.6)
• Anaphylactic reactions in patients with the aspirin triad or in patients without prior exposure to Dyloject: Discontinue Dyloject immediately if an anaphylactic reaction occurs. ( 5.7, 5.13)
• Serious skin reactions such as exfoliative dermatitis, Stevens-Johnson Syndrome (SJS), and toxic epidermal necrolysis (TEN), which can be fatal. Discontinue Dyloject if rash or other signs of local skin reaction occur. ( 5.8)
ADVERSE REACTIONS
The most common adverse reactions (>5%) in controlled clinical trials include nausea, constipation, headache, infusion site pain, dizziness, flatulence, vomiting, and insomnia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Hospira, Inc. at 1-800-441-4100 or electronically at ProductComplaintsPP@hospira.com, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
• Concomitant use of diclofenac and aspirin is not generally recommended because of the potential of increased adverse effects including increased GI bleeding. ( 7.1)
• Concomitant use of Dyloject and anticoagulants has a risk of serious GI bleeding higher than users of either drug alone. ( 7.2)
• ACE inhibitors: NSAIDs may diminish the antihypertensive effect of ACE inhibitors. ( 7.3)
USE IN SPECIFIC POPULATIONS
• Pregnancy: Avoid use after 30 weeks gestation because premature closure of the ductus arteriosus in the fetus may occur. ( 8.1)
• Nursing Mothers: Use with caution as diclofenac may be present in human milk. ( 8.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2014
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
Dyloject is an NSAID indicated in adults for the management of mild to moderate pain and management of moderate to severe pain alone or in combination with opioid analgesics.
2 DOSAGE AND ADMINISTRATION
Use for the shortest duration consistent with individual patient treatment goals [see Warnings and Precautions (5)].
For intravenous administration only.
For the treatment of acute pain, the recommended dose of Dyloject is 37.5 mg administered by intravenous bolus injection over 15 seconds every 6 hours as needed, not to exceed 150 mg/day.
To reduce the risk of renal adverse reactions, patients must be well hydrated prior to administration of Dyloject.
Visually inspect parenteral drug products for particulate matter and discoloration prior to administration. If visibly opaque particles, discoloration or other foreign particles are observed, the solution should not be used.
3 DOSAGE FORMS AND STRENGTHS
Dyloject Injection is available as a 37.5 mg/mL single-dose glass vial.
4 CONTRAINDICATIONS
Dyloject is contraindicated in patients with:
• known hypersensitivity (e.g., anaphylactic reactions and serious skin reactions) to diclofenac [ see Warnings and Precautions (5.7, 5.8)].
• a history of asthma, urticaria, or allergic-type reactions after taking aspirin or other NSAIDs. Severe, rarely fatal anaphylactic-like reactions to NSAIDs have been reported in such patients [ see Warnings and Precautions (5.7, 5.13)].
• perioperative pain in the setting of coronary artery bypass graft (CABG) surgery [ see Warnings and Precautions (5.1)].
• moderate to severe renal insufficiency in the perioperative period and who are at risk for volume depletion [ see Warnings and Precautions (5.3)].
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Thrombotic Events
Clinical trials of several COX-2 selective and nonselective NSAIDs of up to three years duration have shown an increased risk of serious cardiovascular (CV) thrombotic events, myocardial infarction and stroke, which can be fatal. All NSAIDs, both COX-2 selective and nonselective, may have a similar risk. Patients with known CV disease or risk factors for CV disease may be at greater risk. To minimize the potential risk for an adverse CV event in patients treated with an NSAID, use the lowest effective dose for the shortest duration possible. Physicians and patients should remain alert for the development of such events, even in the absence of previous CV symptoms. Patients should be informed about the signs and/or symptoms of serious CV events and the steps to take if they occur.
Two large, controlled clinical trials of a COX-2 selective NSAID for the treatment of pain in the first 10-14 days following CABG surgery found an increased incidence of myocardial infarction and stroke [see Contraindications (4)].
There is no consistent evidence that concurrent use of aspirin mitigates the increased risk of serious CV thrombotic events associated with NSAID use. The concurrent use of aspirin and an NSAID does increase the risk of serious gastrointestinal (GI) events [see Warnings and Precautions (5.2)].
5.2 Gastrointestinal Effects: Risk of Ulceration, Bleeding, and Perforation
NSAIDs, including Dyloject, can cause serious GI adverse events including inflammation, bleeding, ulceration, and perforation of the stomach, small intestine, or large intestine, which can be fatal. These serious adverse events can occur at any time, with or without warning symptoms, in patients treated with NSAIDs. Only one in five patients who develop a serious upper GI adverse event on NSAID therapy is symptomatic. Upper GI ulcers, gross bleeding, or perforation caused by NSAIDs occur in approximately 1% of patients treated for 3-6 months and in about 2-4% of patients treated for one year. These trends continue with longer duration of use, increasing the likelihood of developing a serious GI event at some time during the course of therapy. Dyloject is administered by intravenous injection and is intended for acute short term use. However, even short-term therapy is not without risk.
Prescribe NSAIDs, including Dyloject, with extreme caution in those with a prior history of ulcer disease or GI bleeding. Patients with a prior history of peptic ulcer disease and/or GI bleeding who use NSAIDs have a greater than 10-fold increased risk for developing a GI bleed compared to treated patients with neither of these risk factors. Other factors that increase the risk for GI bleeding in patients treated with NSAIDs include concomitant use of oral corticosteroids or anticoagulants, longer duration of NSAID therapy, smoking, use of alcohol, older age, and poor general health status. Most reports of spontaneous fatal GI events are in elderly or debilitated patients, and therefore special care should be taken in treating this population.
To minimize the potential risk for an adverse GI event in patients treated with an NSAID, use the lowest effective dose for the shortest possible duration. Patients and physicians should remain alert for signs and symptoms of GI ulcerations and bleeding during NSAID therapy and promptly initiate additional evaluation and treatment if a serious GI event is suspected. This should include discontinuation of the NSAID until a serious GI adverse event is ruled out. For high risk patients, alternate therapies that do not involve NSAIDs should be considered.
5.3 Renal Effects
Use caution when initiating treatment with Dyloject in patients with considerable dehydration. Dyloject is not recommended in patients with moderate to severe renal insufficiency and is contraindicated in patients with moderate to severe renal insufficiency in the perioperative period and who are at risk for volume depletion. Acute renal decompensation was observed in 4% out of 68 patients enrolled with renal impairment and treated with Dyloject in clinical trials in the perioperative period.
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other renal injury. Renal toxicity has also been seen in patients in whom renal prostaglandins have a compensatory role in the maintenance of renal perfusion. In these patients, administration of an NSAID may cause a dose dependent reduction in prostaglandin formation and, secondarily, in renal blood flow, which may precipitate overt renal decompensation. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics and ACE inhibitors, and the elderly. Discontinuation of NSAID therapy is usually followed by recovery to the pretreatment state.
5.4 Hepatic Effects
Elevations of one or more liver tests may occur during therapy with Dyloject. These laboratory abnormalities may progress, may remain unchanged, or may be transient with continued therapy. Borderline elevations (i.e., less than 3 times the ULN [ULN = the upper limit of the normal range]) or greater elevations of transaminases occurred in about 15% of diclofenac-treated patients in clinical trials of indications other than acute pain. Of the markers of hepatic function, ALT (SGPT) is recommended for the monitoring of liver injury.
In clinical trials of oral diclofenac, meaningful elevations (i.e., more than 3 times the ULN) of AST (SGOT) occurred in about 2% of approximately 5,700 patients at some time during diclofenac treatment (ALT was not measured in all studies).
In a large, open-label, controlled trial of 3,700 patients treated for 2-6 months, patients were monitored first at 8 weeks and 1,200 patients were monitored again at 24 weeks. Meaningful elevations of ALT and/or AST occurred in about 4% of the 3,700 patients and included marked elevations (i.e., more than 8 times the ULN) in about 1% of the 3,700 patients. In this open-label study, a higher incidence of borderline (less than 3 times the ULN), moderate (3-8 times the ULN), and marked (greater than 8 times the ULN) elevations of ALT or AST was observed in patients receiving diclofenac when compared to other NSAIDs. Elevations in transaminases were seen more frequently in patients with osteoarthritis than in those with rheumatoid arthritis. Almost all meaningful elevations in transaminases were detected before patients became symptomatic. Abnormal tests occurred during the first 2 months of therapy with diclofenac in 42 of the 51 patients in all trials who developed marked transaminase elevations.
In postmarketing reports, cases of drug-induced hepatotoxicity have been reported in the first month, and in some cases, the first 2 months of therapy, but can occur at any time during treatment with diclofenac. Postmarketing surveillance has reported cases of severe hepatic reactions, including liver necrosis, jaundice, fulminant hepatitis with and without jaundice, and liver failure. Some of these reported cases resulted in fatalities or liver transplantation.
Measure transaminases (ALT and AST) periodically in patients receiving long-term therapy with diclofenac, because severe hepatotoxicity may develop without a prodrome of distinguishing symptoms. The optimum times for making the first and subsequent transaminase measurements are not known. Based on clinical trial data and postmarketing experiences, transaminases should be monitored within 4 to 8 weeks after initiating treatment with diclofenac. Dyloject is not indicated for long-term treatment. However, severe hepatic reactions can occur at any time during treatment with diclofenac.
If abnormal liver tests persist or worsen, if clinical signs and/or symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g. eosinophilia, rash, abdominal pain, diarrhea, dark urine, etc.), discontinue Dyloject immediately. To minimize the possibility that hepatic injury will become severe between transaminase measurements, inform patients of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, diarrhea, pruritus, jaundice, right upper quadrant tenderness, and “flu-like” symptoms), and the appropriate action patients should take if these signs and symptoms appear. To minimize the potential risk for an adverse liver-related event in patients treated with diclofenac, use the lowest effective dose for the shortest duration possible. Exercise caution when prescribing Dyloject with concomitant drugs that are known to be potentially hepatotoxic (e.g., acetaminophen, certain antibiotics, anti-epileptics).
5.5 Hypertension
NSAIDs, including Dyloject, can lead to onset of new hypertension or worsening of pre-existing hypertension, either of which may contribute to the increased incidence of CV events. Use NSAIDs, including Dyloject, with caution in patients with hypertension. Monitor blood pressure closely during the initiation of NSAID treatment and throughout the course of therapy.
Patients taking ACE inhibitors, thiazides or loop diuretics may have impaired response to these therapies when taking NSAIDs.
5.6 Congestive Heart Failure and Edema
Fluid retention and edema have been observed in some patients taking NSAIDs. Use Dyloject with caution in patients with fluid retention or heart failure.
5.7 Anaphylactic Reactions
As with other NSAIDs, anaphylactic reactions may occur in patients without known prior exposure to Dyloject. Dyloject is contraindicated in patients with the aspirin triad. This symptom complex typically occurs in asthmatic patients who experience rhinitis with or without nasal polyps, or who exhibit severe, potentially fatal bronchospasm after taking aspirin or other NSAIDs [see Contraindications (4)].
5.8 Serious Skin Reactions
NSAIDs, including Dyloject, can cause serious skin adverse reactions such as exfoliative dermatitis, Stevens-Johnson Syndrome (SJS), and toxic epidermal necrolysis (TEN), which can be fatal. These serious events may occur without warning. Inform patients about the signs and symptoms of serious skin manifestations, and discontinue Dyloject at the first appearance of skin rash or any other sign of hypersensitivity [see Contraindications (4)].
5.9 Pregnancy
Starting at 30 weeks gestation, Dyloject and other NSAIDs, should be avoided by pregnant women as premature closure of the ductus arteriosus in the fetus may occur. If this drug is used during this time period in pregnancy, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations (8.1)].
5.10 Corticosteroid Treatment
Dyloject cannot be expected to substitute for corticosteroids or to treat corticosteroid insufficiency. Abrupt discontinuation of corticosteroids may lead to exacerbation of corticosteroid-responsive illness. Patients on prolonged corticosteroid therapy should have their therapy tapered slowly if a decision is made to discontinue corticosteroids.
5.11 Masking Inflammation and Fever
The pharmacological activity of Dyloject in reducing inflammation, and possibly fever, may diminish the utility of these diagnostic signs in detecting infectious complications of presumed noninfectious, painful conditions.
5.12 Hematological Effects
Anemia may occur in patients receiving NSAIDs, including Dyloject. This may be due to fluid retention, occult or gross GI blood loss, or an incompletely described effect upon erythropoiesis. In patients on long-term treatment with NSAIDs, including diclofenac, check hemoglobin or hematocrit if they exhibit any signs or symptoms of anemia or blood loss. Dyloject is not indicated for long-term treatment.
NSAIDs inhibit platelet aggregation and have been shown to prolong bleeding time in some patients. Unlike aspirin, their effect on platelet function is quantitatively less, of shorter duration, and reversible. Carefully monitor patients who may be adversely affected by alterations in platelet function, such as those with coagulation disorders or patients receiving anticoagulants.
5.13 Pre-existing Asthma
Patients with asthma may have aspirin-sensitive asthma. The use of aspirin in patients with aspirin-sensitive asthma has been associated with severe bronchospasm, which can be fatal. Since cross-reactivity between aspirin and NSAIDs has been reported in such aspirin-sensitive patients, including bronchospasm, Dyloject is contraindicated in patients with this form of aspirin sensitivity and should be used with caution in all patients with pre-existing asthma [see Contraindications (4)].
5.14 Monitoring
Because serious GI tract ulcerations and bleeding can occur without warning symptoms, monitor for signs or symptoms of GI bleeding.
For patients on long-term treatment with NSAIDs, periodically check a CBC and chemistry profile, including liver function tests. Discontinue Dyloject if clinical signs and symptoms consistent with liver or renal disease develop, systemic manifestations occur (e.g., eosinophilia, rash), or abnormal liver tests persist or worsen. Dyloject is not indicated for long-term treatment.
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in the labeling:
• Cardiovascular thrombotic events [ see Boxed Warning and Warnings and Precautions (5.1)]
• Gastrointestinal effects [ see Boxed Warning and Warnings and Precautions (5.2)]
• Renal effects [ see Contraindications (4) and Warnings and Precautions (5.3)]
• Hepatic effects [ see Warnings and Precautions (5.4)]
• Hypertension [ see Warnings and Precautions (5.5)]
• Congestive heart failure and edema [ see Warnings and Precautions (5.6)]
• Anaphylactoid reactions [ see Warnings and Precautions (5.7)]
• Serious skin reactions [ see Warnings and Precautions (5.8)]
Adverse reactions from clinical studies of Dyloject
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
During clinical development, 1,156 patients were exposed to Dyloject in multiple-dose, controlled and open-label studies. Dyloject was administered post-surgically every 6 hours for up to 5 days. The incidence rates of adverse reactions listed in the following table are derived from multicenter, controlled clinical studies in post-operative patients comparing Dyloject to placebo in patients who may have also received morphine rescue medication.
Table 1: Proportion of Patients Experiencing Common Adverse Reactions in Placebo-Controlled Clinical Studies in Patients with Acute Moderate-to-Severe Postoperative Pain occurring in greater than or equal to 3% in patients treated with Dyloject*
|
MedDRA Preferred Term |
Placebo
N=126 |
Dyloject
N=187 |
|
Any Reaction |
104 (83%) |
146 (78%) |
|
Nausea
Constipation
Headache
Infusion Site Pain
Dizziness
Flatulence
Vomiting
Insomnia
Pruritus
Hypotension
Pyrexia
Anemia
Infusion Site Extravasation |
50 (40%)
14 (11%)
20 (16%)
10 (8%)
2 (2%)
20 (16%)
23 (18%)
12 (10%)
10 (8%)
6 (5%)
13 (10%)
9 (7%)
1 (1%) |
45 (24%)
25 (13%)
19 (10%)
19 (10%)
15 (8%)
15 (8%)
12 (6%)
11 (6%)
9 (5%)
9 (5%)
8 (4%)
8 (4%)
6 (3%) | Intravenous morphine was permitted as rescue medication for pain management.
Adverse reactions from clinical studies or spontaneous reports for other formulations of diclofenac and other NSAIDs
In patients taking diclofenac or other NSAIDs, the most frequently reported adverse reactions occurring in approximately 1%-10% of patients are:
Gastrointestinal experiences including abdominal pain, constipation, diarrhea, dyspepsia, flatulence, gross bleeding/perforation, heartburn, nausea, GI ulcers (gastric/duodenal) and vomiting.
Abnormal renal function, anemia, dizziness, edema, elevated liver enzymes, headaches, increased bleeding time, pruritus, rashes and tinnitus.
Additional adverse reactions reported occasionally include:
Body as a Whole: fever, infection, sepsis
Cardiovascular System: congestive heart failure, hypertension, tachycardia, syncope
Digestive System: esophagitis, gastric/peptic ulcers, gastritis, gastrointestinal bleeding, glossitis, hematemesis, hepatitis, jaundice
Hemic and Lymphatic System: ecchymosis, eosinophilia, leukopenia, melena, purpura, rectal bleeding, stomatitis, thrombocytopenia
Metabolic and Nutritional: weight changes
Nervous System: anxiety, asthenia, confusion, depression, dream abnormalities, drowsiness, insomnia, malaise, nervousness, paresthesia, somnolence, tremors, vertigo
Respiratory System: asthma, dyspnea
Skin and Appendages: alopecia, photosensitivity, sweating increased
Special Senses: blurred vision
Urogenital System: cystitis, dysuria, hematuria, interstitial nephritis, oliguria/polyuria, proteinuria, renal failure
Other adverse reactions, which occur rarely are:
Body as a Whole: anaphylactic reactions, appetite changes, death
Cardiovascular System: arrhythmia, hypotension, myocardial infarction, palpitations, vasculitis
Digestive System: colitis, eructation, fulminant hepatitis with and without jaundice, liver failure, liver necrosis, pancreatitis
Hemic and Lymphatic System: agranulocytosis, hemolytic anemia, aplastic anemia, lymphadenopathy, pancytopenia
Metabolic and Nutritional: hyperglycemia
Nervous System: convulsions, coma, hallucinations, meningitis
Respiratory System: respiratory depression, pneumonia
Skin and Appendages: angioedema, toxic epidermal necrolysis, erythema multiforme, exfoliative dermatitis, Stevens-Johnson syndrome, urticaria
Special Senses: conjunctivitis, hearing impairment
Adverse reactions of special interest
Based on the analysis of the pooled data from the multi-dose, controlled clinical trials, post-operative patients treated with Dyloject had more adverse reactions related to wound healing (7.5%) compared to patients treated with placebo (4%).
7 DRUG INTERACTIONS
7.1 Aspirin
When administered with aspirin, the protein binding of Dyloject is reduced. The clinical significance of this interaction is not known; however, as with other NSAIDs, concomitant administration of Dyloject and aspirin is not generally recommended because of the potential of increased adverse effects.
7.2 Anticoagulants
The effects of anticoagulants (e.g., warfarin) and NSAIDs on GI bleeding are synergistic, such that the users of both drugs together have a higher risk of serious GI bleeding than users of either drug alone [see Warnings and Precautions (5.2)].
7.3 ACE Inhibitors
NSAIDs may diminish the antihypertensive effect of ACE inhibitors. This interaction should be given consideration in patients taking NSAIDs concomitantly with ACE inhibitors.
7.4 Cyclosporine
NSAIDs, including Dyloject, may affect renal prostaglandins and increase the toxicity of certain drugs. Therefore, concomitant therapy with Dyloject may increase cyclosporine’s nephrotoxicity. Use caution when Dyloject is administered concomitantly with cyclosporine.
7.5 Diuretics
Clinical studies and postmarketing observations have shown that Dyloject can reduce the natriuretic effect of furosemide and thiazides in some patients. This response has been attributed to inhibition of renal prostaglandin synthesis. During concomitant therapy with NSAIDs, observe patients closely for signs of renal failure, as well as to assure diuretic efficacy [see Warnings and Precautions (5.3, 5.6)].
7.6 Lithium
NSAIDs have produced elevations of plasma lithium levels and a reduction in renal lithium clearance. The mean minimum lithium concentration increased 15% and the renal clearance decreased by 20%. This effect has been attributed to inhibition of renal prostaglandin synthesis by the NSAID. Thus, when NSAIDs and lithium are administered concurrently, observe patients carefully for signs of lithium toxicity.
7.7 Methotrexate
NSAIDs have been reported to competitively inhibit methotrexate accumulation in rabbit kidney slices. This indicates that NSAIDs may enhance the toxicity of methotrexate. Use caution when NSAIDs are administered concomitantly with methotrexate.
7.8 CYP2C9 Inhibitors or Inducers
Diclofenac is metabolized by cytochrome P450 enzymes, predominantly by CYP2C9. Co‑administration of diclofenac with CYP2C9 inhibitors (e.g. voriconazole) may enhance the exposure and toxicity of diclofenac whereas co-administration with CYP2C9 inducers (e.g. rifampin) may lead to compromised efficacy of diclofenac. Use caution when dosing Dyloject with CYP2C9 inhibitors or inducers; a dosage adjustment may be warranted.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Teratogenic Effects - Pregnancy Category C prior to 30 weeks gestation; Category D starting at 30 weeks gestation.
Starting at 30 weeks gestation, Dyloject and other NSAIDs should be avoided by pregnant women as premature closure of the ductus arteriosus in the fetus may occur. Dyloject can cause fetal harm when administered to a pregnant woman starting at 30 weeks gestation.
There are no adequate and well-controlled studies in pregnant women. Prior to 30 weeks gestation, Dyloject should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Reproductive studies have been performed in mice given diclofenac sodium (up to 20 mg/kg/day or 60 mg/m2/day) and in rats and rabbits given diclofenac sodium (up to 10 mg/kg/day or 60 mg/m2/day for rats, and 80 mg/m2/day for rabbits, 0.75-fold and 1-fold the maximum recommended human dose [MRHD] of 150 mg/day based on body surface area comparison, respectively), and have revealed no evidence of teratogenicity despite the induction of maternal toxicity and fetal toxicity. In rats maternally toxic doses were associated with dystocia, prolonged gestation, reduced fetal weights and growth, and reduced fetal survival. Diclofenac has been shown to cross the placental barrier in mice, rats, and humans.
8.2 Labor and Delivery
Dyloject should not be used in labor and delivery due to the inhibitory effects on prostaglandin synthesis that may adversely affect fetal circulation, inhibit uterine contractions and increase risk of uterine bleeding.
In rat studies, maternal exposure to NSAIDs, as with other drugs known to inhibit prostaglandin synthesis, increased the incidence of dystocia and delayed parturition, and decreased pup survival.
8.3 Nursing Mothers
Based on available data, diclofenac may be present in human milk. One woman treated orally with a diclofenac salt, 150 mg/day, had a milk diclofenac level of 100 mcg/L, equivalent to an infant dose of about 0.03 mg/kg/day. Diclofenac was not detectable in breast milk in 12 women using diclofenac (after either 100 mg/day orally for 7 days or a single 50 mg intramuscular dose administered in the immediate postpartum period). Exercise caution when Dyloject is administered to a nursing woman.
8.4 Pediatric Use
The safety and efficacy of Dyloject have not been established in pediatric patients.
8.5 Geriatric Use
Use caution in elderly patients given the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Diclofenac metabolites are known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, use caution in this patient population, and it may be useful to monitor renal function.
Elderly patients are at increased risk for serious GI and renal adverse events. Older age increases the risk for GI bleeding. Most spontaneous reports of fatal GI events are in elderly or debilitated patients, and therefore special care should be taken in treating this population [see Warnings and Precautions (5.2)].
The pharmacokinetics of Dyloject are similar in elderly compared to young adults [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
Orally administered diclofenac sodium is extensively metabolized. The pharmacokinetics of Dyloject are similar in patients with mild hepatic impairment compared to healthy subjects [see Clinical Pharmacology (12.3)]. Dosing adjustments in patients with mild hepatic impairment is not necessary. The pharmacokinetics of Dyloject were not studied in patients with moderate to severe hepatic impairment and use in this population is not recommended.
8.7 Renal Impairment
Pharmacokinetics of Dyloject in patients with mild to moderate renal impairment is similar compared to healthy subjects. However, acute renal decompensation was observed in 4% out of 68 patients enrolled with renal impairment and treated with Dyloject in clinical trials in the perioperative period. Dyloject is not recommended in patients with moderate to severe renal insufficiency and is contraindicated in patients with moderate to severe renal insufficiency in the perioperative period and who are at risk for volume depletion [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
8.8 Body Weight
Pharmacokinetics of diclofenac following Dyloject injection appear to be dependent on body weight. The effect of body weight on clinical efficacy and safety of Dyloject has not been fully studied. Therefore, adjusting dose based on body weight is not recommended [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
Symptoms following acute NSAID overdoses are usually limited to lethargy, drowsiness, nausea, vomiting, and epigastric pain, which are generally reversible with supportive care. Gastrointestinal bleeding can occur. Hypertension, acute renal failure, respiratory depression and coma may occur, but are rare. Anaphylactoid reactions have been reported with therapeutic ingestion of NSAIDs, and may occur following an overdose.
Patients should be managed by symptomatic and supportive care following an NSAID overdose. There are no specific antidotes. Forced diuresis, alkalinization of urine, hemodialysis, or hemoperfusion may be employed but are not likely to be useful due to high protein binding. In case of an overdosage, discontinue Dyloject therapy and consider contacting a regional poison control center at 1-800-222-1222.
11 DESCRIPTION
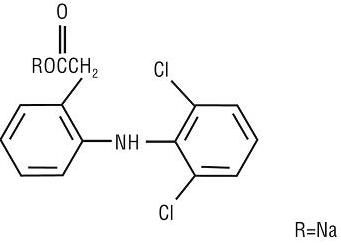
Dyloject Injection is a clear, colorless solution for intravenous administration. Each mL of aqueous solution contains 37.5 mg of diclofenac sodium (34.8 mg of diclofenac), 333 mg hydroxypropyl betadex (HPβCD), 5 mg monothioglycerol, sodium hydroxide and/or hydrochloric acid for pH adjustment, and water for injection.
Diclofenac sodium, a nonsteroidal anti-inflammatory drug, is designated chemically as benzeneacetic acid, 2-[(2,6-dichlorophenyl)amino] monosodium salt, with a molecular formula of C14H10Cl2NNaO2. The molecular weight is 318.13. The structural formula of diclofenac sodium is:
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The active ingredient in Dyloject, diclofenac, is an NSAID that exhibits anti-inflammatory analgesic and antipyretic activities in animal models.
The mechanism of action of Dyloject, like that of other NSAIDs, is not completely understood but may involve inhibition of the cyclooxygenase (COX-1 and COX-2) pathways. Dyloject’s mechanism may also be related to inhibition of prostaglandin synthetase.
12.2 Pharmacodynamics
The effect of Dyloject on QTc prolongation was evaluated in a randomized, double-blind, positive (moxifloxacin 400 mg) - and placebo-controlled crossover study in healthy subjects. A total of 70 subjects was administered diclofenac sodium 37.5 mg and 75 mg. In a study with demonstrated ability to detect small effects, the upper bound of the 90% confidence interval for the largest placebo-adjusted, baseline-corrected QTc based on Fridericia correction method (QTcF) was below 10 ms, the threshold for regulatory concern.
12.3 Pharmacokinetics
Following intravenous administration of Dyloject to healthy volunteers, plasma concentrations of diclofenac exceed that of immediate-release oral diclofenac for the first 45 minutes reaching a maximum of 4.8-fold 5 minutes after administration.
The pharmacokinetics of diclofenac following intravenous administration of Dyloject and oral doses of immediate-release diclofenac are compared in Table 2.
Table 2: Single-dose and Multiple-dose Pharmacokinetics of Dyloject (diclofenac sodium) Injection and Oral Immediate Release (IR) Diclofenac Potassium
|
Parameter1 |
Dyloject
37.5 mg
IV |
Oral IR Diclofenac
50 mg
PO |
|
Single Dose |
|
Cmax (ng/mL)
Tmax (h)
AUC(inf) (h·ng/mL)
t1/2 (h)
CL (mL/min)
Vz (L) |
6,031 ± 1,178
0.083
1,859 ± 376
1.44 ± 0.27
324 ± 63.0
40.1 ± 9.77 |
1,246 ±732
1.5
1,562 ± 519
1.28 ± 0.27
526 ± 179
57.3 ± 20.4 |
|
Multiple Dose |
|
Cmax (ng/mL)
Tmax (h)
AUC(0-t) (h·ng/mL)
t1/2 (h)
CL (mL/min)
Vz (L) |
5,617 ± 1,799
0.083
1,839 ± 506
2.29 ± 0.63
387 ± 394
83.4 ± 127 |
851 ± 462
1.49
1,350 ± 601
2.80 ± 0.66
894 ± 1,392
242 ± 486 | IV=intravenous; PO=oral; 1CL and Vz are CL/F and Vz/F for oral immediate release diclofenac
Dyloject administered as an intravenous bolus dose of 37.5 mg every 6 hours for 4 doses to healthy subjects (N=36) showed minimal accumulation with mean values for Cmax and AUC equivalent between the first and the fourth dose.
Dyloject exhibits linear pharmacokinetics over intravenous doses ranging from 18.75 to 75 mg and injection times ranging from a bolus (less than 5 seconds) to 60 seconds.
Distribution
Following administration of Dyloject, the apparent volume of distribution during the terminal elimination phase (Vz) of diclofenac is 40.1 ± 9.77 L.
Diclofenac is more than 99% bound to human serum proteins, primarily albumin. Serum binding is constant over the concentration range (0.15-105 mcg/mL) achieved with the recommended doses.
Diclofenac diffuses into and out of the synovial fluid. Diffusion into the joint occurs when plasma levels are higher than those in the synovial fluid, after which the process reverses and synovial fluid levels are higher than plasma levels. It is not known whether diffusion into the joint plays a role in the effectiveness of diclofenac.
HPβCD is distributed in the extracellular fluids following administration of Dyloject, and has a volume of distribution during the terminal elimination phase (Vz) of 21.8 ±7.36 L.
Metabolism
Five diclofenac metabolites have been identified in human plasma and urine. The metabolites include 4’-hydroxy-, 5-hydroxy-, 3’-hydroxy-, 4’, 5 -dihydroxy- and 3’-hydroxy-4’-methoxy diclofenac. The major diclofenac metabolite, 4’-hydroxy-diclofenac, has very weak pharmacologic activity. The formation of 4’-hydroxy diclofenac is primarily mediated by CYP2C9. Both diclofenac and its oxidative metabolites undergo glucuronidation or sulfation followed by biliary excretion. Acylglucuronidation mediated by UGT2B7 and oxidation mediated by CYP2C8 may also play a role in diclofenac metabolism. CYP3A4 is responsible for the formation of minor metabolites, 5-hydroxy and 3’-hydroxy- diclofenac.
In patients with renal dysfunction, peak concentrations of metabolites 4’-hydroxy- and 5-hydroxy-diclofenac were approximately 50% and 4% of the parent compound after single oral dosing compared to 27% and 1% in normal healthy subjects.
Excretion
Diclofenac is eliminated through metabolism and subsequent urinary and biliary excretion of the glucuronide and the sulfate conjugates of the metabolites.
Plasma concentrations of Dyloject decline from peak levels in a biexponential fashion, with a terminal phase half-life of approximately 1.4 hours following intravenous administration.
Total systemic clearance of diclofenac in plasma following administration of Dyloject is 324 ± 63 mL/min.
Little or no free unchanged diclofenac is excreted in the urine following administration of Dyloject. Approximately 65% of the dose is excreted in the urine and approximately 35% in the bile as conjugates of unchanged diclofenac plus metabolites. Less than 1% is excreted as unchanged substance.
The terminal half-life of HPβCD in plasma following administration of Dyloject is approximately 2.7 ± 1.4 hours.
Special Populations
Age
Pediatric: The pharmacokinetics of Dyloject have not been established in pediatric subjects [see Use in Specific Populations (8.4)].
Geriatrics: The effect of aging on the pharmacokinetics of Dyloject was studied in 88 subjects from 18 to 86 years old. The terminal half-life for subjects aged 65 to 74 years was 1.4 hours and for subjects greater than or equal to 75 years was 2.1 hours. Clearance of diclofenac following administration of Dyloject was not affected by age [see Use in Specific Populations (8.5)].
Race: Pharmacokinetics of diclofenac following injection of Dyloject was studied in Caucasian, Black/African and Asian subjects. After taking body weight into account there was no difference in pharmacokinetics of diclofenac with respect to race [see Pharmacokinetics (12.3)].
Gender: Systemic exposure of diclofenac was 30% higher in females compared to males following Dyloject administration. However, this is possibly due to the effect of body weight on clearance of diclofenac. After taking body weight into account there was no difference in pharmacokinetics of diclofenac with respect to gender [see Pharmacokinetics (12.3)].
Hepatic Insufficiency
The pharmacokinetics of Dyloject were evaluated in 8 subjects with mild hepatic impairment (Child-Pugh Classification A, Score of 5 to 6 and a bilirubin of less than or equal to 2.5 mg/dL) compared to matched healthy subjects. The pharmacokinetics of diclofenac following administration of Dyloject in mild hepatic impaired subjects were not altered. Pharmacokinetics of Dyloject has not been evaluated in moderate or severe hepatic impaired subjects [see Use in Specific Populations (8.6)].
Renal Insufficiency
The pharmacokinetics of Dyloject in mild (n = 8), and moderate (n = 5) renal impaired subjects were not significantly altered compared to healthy subjects (n = 7) [see Warnings and Precautions (5.3) and Use in Specific Populations (8.7)].
Effect of Body Weight
Pharmacokinetics of diclofenac following Dyloject injection appear to be dependent on body weight. The pharmacokinetics of Dyloject were studied in 88 subjects ranging in weight from 53 to 156.2 kg. Clearance of diclofenac in subjects weighing below 95 kg is 282±68 mL/min compared to 356±53 mL/min in subjects above 95 kg body weight (approximately 30% higher clearance). The volume of distribution increased with increased body weight and the proportional increase in clearance resulted in no change in elimination half-life with increased body weight [see Use in Specific Populations (8.8)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: Long-term carcinogenicity studies in rats given diclofenac sodium up to 2 mg/kg/day (0.13-fold the maximum recommended human dose [MRHD] of 150 mg/day based on mg/m2 body surface area [BSA] comparison) have revealed no significant increase in tumor incidence. A 2-year carcinogenicity study conducted in mice employing diclofenac sodium at doses up to 0.03 mg/kg/day (0.01-fold the MRHD of 150 mg/day based on BSA comparison) in males and 1 mg/kg/day (0.04-fold the MRHD of 150 mg/day based on BSA comparison) in females did not reveal any oncogenic potential.
Mutagenesis: Diclofenac sodium did not show mutagenic activity in in vitro point mutation assays in mammalian (mouse lymphoma) and microbial (yeast, Ames) test systems and was nonmutagenic in several mammalian in vitro and in vivo tests, including dominant lethal and male germinal epithelial chromosomal aberration studies in Chinese hamsters.
Impairment of Fertility: Diclofenac sodium administered to male and female rats at 4 mg/kg/day (0.3-fold the MRHD of 150 mg/day based on BSA comparison) did not affect fertility.
14 CLINICAL STUDIES
The effect of Dyloject in the short-term treatment of acute pain was evaluated in two double-blind, placebo and active-controlled, multiple-dose clinical trials in patients with postoperative pain. In both trials, intravenous morphine was permitted as rescue medication for pain management.
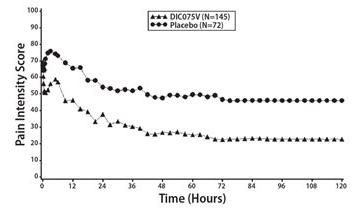
In a controlled, multiple-dose study of adult patients with postoperative pain who had undergone elective abdominal or pelvic surgery, 245 patients were treated with Dyloject, a positive NSAID control (ketorolac tromethamine), or placebo administered every 6 hours starting within 6 hours after surgery and for up to 5 days. The study population consisted of patients with a mean age of 43 years (range 18 to 65 years) and a minimum pain intensity of 50 mm on a 100-mm visual analog scale (VAS) at baseline. The mean baseline pain intensity on the VAS was 68 mm (range 50 to 100 mm). Approximately 63% of subjects in the Dyloject 37.5 mg group and 92% of subjects in the placebo group took rescue medication within the first 48 hours of the treatment phase. Efficacy was demonstrated by a reduction in pain intensity as measured by the sum of the pain intensity differences over 0 to 48 hours in patients receiving Dyloject as compared to placebo. The average pain intensities over time are depicted for the treatment groups in Figure 1.
In a second controlled, multiple-dose study of adult patients with postoperative pain who had undergone elective orthopedic surgery, 277 patients were treated with Dyloject, a positive NSAID control (ketorolac tromethamine), or placebo administered every 6 hours starting within 6 hours postsurgery and for up to 5 days. The study population consisted of patients with a mean age of 55 years (range 19 to 84 years) and a minimum pain intensity of 50 mm on a 100-mm VAS at baseline. The mean baseline pain intensity on the VAS was 69 mm (range 50 to 100 mm). Approximately 74% of subjects in the Dyloject group and 92% of subjects in the placebo group took rescue medication within the first 48 hours of the treatment phase. Efficacy was demonstrated by a reduction in pain intensity as measured by the sum of the pain intensity differences over 0 to 48 hours in patients receiving Dyloject as compared to placebo. The average pain intensities over time are depicted for the treatment groups in Figure 2.
Figure 1: Pain Intensity Score Versus Time
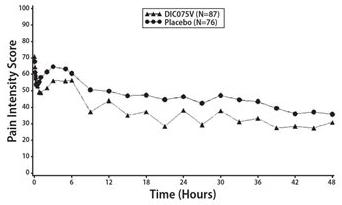
Figure 2: Pain Intensity Score Versus Time
16 HOW SUPPLIED/STORAGE AND HANDLING
Dyloject (diclofenac sodium) Injection, is a clear, colorless solution that is supplied as a 1 mL fill in a clear 2 mL glass vial with an orange tamper-evident top.
|
NDC No. |
Strength |
Fill |
Container Size |
|
0409-1068-01 |
37.5 mg/mL |
1 mL |
2 mL | Carton of 25 vials
Store at Controlled Room Temperature 20 to 25°C (68 to 77°F) [see USP Controlled Room Temperature].
Do not freeze. Protect from light.
Keep Dyloject out of reach and sight of children.
Not made with natural rubber latex.
17 PATIENT COUNSELING INFORMATION
Patients should be informed of the following information before initiating therapy with an NSAID and periodically during the course of ongoing therapy. Patients should be informed of the following information before initiating therapy with an NSAID.
Cardiovascular Effects
NSAIDs, including Dyloject, may cause serious CV side effects such as myocardial infarction or stroke, which may result in hospitalization and even death. Although serious CV events can occur without warning symptoms, advise patients to be alert for the signs and symptoms of chest pain, shortness of breath, weakness, and slurring of speech, and to ask for medical advice when observing any indicative sign or symptoms. Inform patients of the importance of this follow-up [see Warnings and Precautions (5.1)].
Gastrointestinal Effects
NSAIDs, including Dyloject, can cause GI discomfort and, rarely, serious GI side effects, such as ulcers and bleeding, which may result in hospitalization and even death. Although serious GI tract ulcerations and bleeding can occur without warning symptoms, advise patients to be alert for the signs and symptoms of ulcerations and bleeding, and to ask for medical advice when observing any indicative sign or symptoms including epigastric pain, dyspepsia, melena, and hematemesis. Inform patients of the importance of this follow-up [see Warnings and Precautions (5.2)].
Hepatotoxicity
Inform patients of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, diarrhea, pruritus, jaundice, right upper quadrant tenderness, and “flu-like” symptoms). Instruct patients to stop therapy with Dyloject and seek immediate medical therapy if any of these occur [see Warnings and Precautions (5.4)].
Serious Skin Reactions
NSAIDs, including Dyloject, can cause serious skin reactions such as exfoliative dermatitis, SJS, and TEN, which may result in hospitalization and even death. Although serious skin reactions may occur without warning, patients should be alert for the signs and symptoms of skin rash and blisters, fever, or other signs of hypersensitivity such as itching, and to ask for medical advice when observing any indicative sign or symptoms. Advise patients to stop Dyloject immediately if they develop any type of rash, and to contact a physician as soon as possible [see Warnings and Precautions (5.8)].
Weight Gain and Edema
Advise patients to promptly report to their physicians signs or symptoms of unexplained weight gain or edema during treatment with Dyloject [see Warnings and Precautions (5.6)].
Anaphylactic Reactions
Inform patients of the signs and symptoms of an anaphylactic reaction (e.g., difficulty breathing, swelling of the face or throat). If these occur, therapy should be discontinued and medical therapy initiated [see Warnings and Precautions (5.7)].
Effects During Pregnancy
Starting at 30 weeks gestation, Dyloject and other NSAIDs should be avoided by pregnant women as premature closure of the ductus arteriosus in the fetus may occur [see Use in Specific Populations (8.1)]. |
