|
新型抗癌药YONDELIS(TRABECTEDIN/POWDER IV (INFUSION)ET-743)获美国FDA扩展治疗晚期软组织肉瘤
YONDELIS is an alkylating drug indicated for the treatment of patients with unresectable or metastatic liposarcoma or leiomyosarcoma who received a prior anthracycline-containing regimen (1) DOSAGE AND ADMINISTRATION Administer at 1.5 mg/m2 body surface area as a 24-hour intravenous infusion, every 3 weeks through a central venous line (2.1, 2.5) Premedication: dexamethasone 20 mg IV, 30 min before each infusion (2.2) Hepatic Impairment: Administer at 0.9 mg/m2 body surface area as a 24-hour intravenous infusion, every 3 weeks through a central venous line in patients with moderate hepatic impairment (2.1, 5.3, 8.6, 12.3) DOSAGE FORMS AND STRENGTHS For injection: 1 mg sterile lyophilized powder in a single-dose vial (3) CONTRAINDICATIONS Known hypersensitivity to trabectedin (4) WARNINGS AND PRECAUTIONS Neutropenic sepsis: Severe, and fatal, neutropenic sepsis may occur. Monitor neutrophil count during treatment. Withhold YONDELIS for Grade 2 or greater neutropenia (5.1) Rhabdomyolysis: Rhabdomyolysis may occur; withhold YONDELIS for severe or life-threatening increases in creatine phosphokinase level (5.2) Hepatotoxicity: Hepatotoxicity may occur. Monitor and delay and/or reduce dose if needed (5.3) Cardiomyopathy: Severe and fatal cardiomyopathy can occur. Withhold YONDELIS in patients with left ventricular dysfunction (5.4) Embryofetal toxicity: Can cause fetal harm. Advise of potential risk to a fetus and use effective contraception (5.6, 8.1, 8.3) ADVERSE REACTIONS The most common (≥20%) adverse reactions are nausea, fatigue, vomiting, constipation, decreased appetite, diarrhea, peripheral edema, dyspnea, and headache. The most common (≥5%) grades 3–4 laboratory abnormalities are: neutropenia, increased ALT, thrombocytopenia, anemia, increased AST, and increased creatine phosphokinase. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Janssen Biotech, Inc. at 1-800-526-7736 (1-800-JANSSEN) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS CYP3A inhibitors: Avoid concomitant strong CYP3A inhibitors (7.1) CYP3A inducers: Avoid concomitant strong CYP3A inducers (7.2) USE IN SPECIFIC POPULATIONS Lactation: Breastfeeding not recommended (8.2) Do not administer YONDELIS to patients with severe hepatic impairment (5.3, 8.6, 12.3) See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 7/2016 FULL PRESCRIBING INFORMATION: CONTENTS* 1 INDICATIONS AND USAGE YONDELIS® is indicated for the treatment of patients with unresectable or metastatic liposarcoma or leiomyosarcoma who received a prior anthracycline-containing regimen [see Clinical Studies (14)]. 2 DOSAGE AND ADMINISTRATION 2.1 Recommended Dose and Schedule The recommended dose is 1.5 mg/m2 administered as an intravenous infusion over 24 hours through a central venous line every 21 days (3 weeks), until disease progression or unacceptable toxicity, in patients with normal bilirubin and AST or ALT less than or equal to 2.5 times the upper limit of normal. Hepatic Impairment: The recommended dose is 0.9 mg/m2 in patients with moderate hepatic impairment (bilirubin levels 1.5 times to 3 times the upper limit of normal, and AST and ALT less than 8 times the upper limit of normal). Do not administer YONDELIS to patients with severe hepatic impairment (bilirubin levels above 3 times to 10 times the upper limit of normal, and any AST and ALT) [see Warnings and Precautions (5.3), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)]. 2.2 Premedication Administer dexamethasone 20 mg intravenously 30 minutes prior to each dose of YONDELIS. 2.3 Dose Modifications Permanently discontinue YONDELIS for: Persistent adverse reactions requiring a delay in dosing of more than 3 weeks. Adverse reactions requiring dose reduction following YONDELIS administered at 1.0 mg/m2 for patients with normal hepatic function or at 0.3 mg/m2 for patients with pre-existing moderate hepatic impairment. Severe liver dysfunction all of the following: bilirubin two times the upper limit of normal and AST or ALT three times the upper limit of normal with alkaline phosphatase less than two times the upper limit of normal in the prior treatment cycle for patients with normal liver function at baseline. Exacerbation of liver dysfunction in patients with pre-existing moderate hepatic impairment. The recommended dose modifications for adverse reactions are listed in Table 1. Once reduced, the dose of YONDELIS should not be increased in subsequent treatment cycles. Table 1: Recommended Dose Modification
The recommended starting doses and dose reductions for YONDELIS are listed in Table 2: Table 2: Recommended Starting Doses and Dose Reductions
2.4 Preparation for Administration YONDELIS is a cytotoxic drug. Follow applicable special handling and disposal procedures.1 Using aseptic technique, inject 20 mL of Sterile Water for Injection, USP into the vial. Shake the vial until complete dissolution. The reconstituted solution is clear, colorless to pale brownish-yellow, and contains 0.05 mg/mL of trabectedin. Inspect for particulate matter and discoloration prior to further dilution. Discard vial if particles or discoloration are observed. Immediately following reconstitution, withdraw the calculated volume of trabectedin and further dilute in 500 mL of 0.9% Sodium Chloride, USP or 5% Dextrose Injection, USP. Do not mix YONDELIS with other drugs. Discard any remaining solution within 30 hours of reconstituting the lyophilized powder. YONDELIS diluted solution is compatible with Type I colorless glass vials, polyvinylchloride (PVC) and polyethylene (PE) bags and tubing, PE and polypropylene (PP) mixture bags, polyethersulfone (PES) in-line filters, titanium, platinum or plastic ports, silicone and polyurethane catheters, and pumps having contact surfaces made of PVC, PE, or PE/PP. 2.5 Administration Infuse the reconstituted, diluted solution over 24 hours through a central venous line using an infusion set with a 0.2 micron polyethersulfone (PES) in-line filter to reduce the risk of exposure to adventitious pathogens that may be introduced during solution preparation. Complete infusion within 30 hours of initial reconstitution. Discard any unused portion of the reconstituted product or of the infusion solution. 3 DOSAGE FORMS AND STRENGTHS For injection: 1 mg, lyophilized powder in single-dose vial for reconstitution. 4 CONTRAINDICATIONS YONDELIS is contraindicated in patients with known severe hypersensitivity, including anaphylaxis, to trabectedin. 5 WARNINGS AND PRECAUTIONS 5.1 Neutropenic Sepsis Neutropenic sepsis, including fatal cases, can occur with YONDELIS. In Trial 1, the incidence of Grade 3 or 4 neutropenia, based on laboratory values, in patients receiving YONDELIS was 43% (161/378). The median time to the first occurrence of Grade 3 or 4 neutropenia was 16 days (range: 8 days to 9.7 months); the median time to complete resolution of neutropenia was 13 days (range: 3 days to 2.3 months). Febrile neutropenia (fever ≥38.5°C with Grade 3 or 4 neutropenia) occurred in 18 patients (5%) treated with YONDELIS. Ten patients (2.6%) experienced neutropenic sepsis, 5 of whom had febrile neutropenia, which was fatal in 4 patients (1.1%). Assess neutrophil count prior to administration of each dose of YONDELIS and periodically throughout the treatment cycle. Withhold YONDELIS for neutrophil counts of less than 1,500 cells/microliter on the day of dosing. Permanently reduce the dose of YONDELIS for life-threatening or prolonged, severe neutropenia in the preceding cycle [see Dosage and Administration (2.3)]. 5.2 Rhabdomyolysis YONDELIS can cause rhabdomyolysis and musculoskeletal toxicity. In Trial 1, rhabdomyolysis leading to death occurred in 3 (0.8%) of the 378 patients receiving YONDELIS. Elevations in creatine phosphokinase (CPK) occurred in 122 (32%) of the 378 patients receiving YONDELIS, including Grade 3 or 4 CPK elevation in 24 patients (6%), compared to 15 (9%) of the 172 patients receiving dacarbazine with any CPK elevation, including 1 patient (0.6%) with Grade 3 CPK elevation. Among the 24 patients receiving YONDELIS with Grade 3 or 4 CPK elevation, renal failure occurred in 11 patients (2.9%); rhabdomyolysis with the complication of renal failure occurred in 4 of these 11 patients (1.1%). The median time to first occurrence of Grade 3 or 4 CPK elevations was 2 months (range: 1 to 11.5 months). The median time to complete resolution was 14 days (range: 5 days to 1 month). Assess CPK levels prior to each administration of YONDELIS. Withhold YONDELIS for serum CPK levels more than 2.5 times the upper limit of normal. Permanently discontinue YONDELIS for rhabdomyolysis [see Dosage and Administration (2.3)]. 5.3 Hepatotoxicity Hepatotoxicity, including hepatic failure, can occur with YONDELIS. Patients with serum bilirubin levels above the upper limit of normal or AST or ALT levels >2.5 × upper limit of normal were not enrolled in Trial 1. In Trial 1, the incidence of Grade 3–4 elevated liver function tests (LFTs; defined as elevations in ALT, AST, total bilirubin, or alkaline phosphatase) was 35% (134/378) in patients receiving YONDELIS. The median time to development of Grade 3–4 elevation in ALT or AST was 29 days (range: 3 days to 11.5 months). Of the 134 patients with Grade 3–4 elevations in LFTs, 114 (85%) experienced complete resolution with the median time to complete resolution of 13 days (range: 4 days to 4.4 months). In Trial 1, the incidence of drug-induced liver injury (defined as concurrent elevation in ALT or AST of more than three times the upper limit of normal, alkaline phosphatase less than two times the upper limit of normal, and total bilirubin at least two times the upper limit of normal) was 1.3% (5/378) in patients receiving YONDELIS. ALT or AST elevation greater than eight times the upper limit of normal occurred in 18% (67/378) of patients receiving YONDELIS. Assess LFTs prior to each administration of YONDELIS and as clinically indicated based on underlying severity of pre-existing hepatic impairment. Manage elevated LFTs with treatment interruption, dose reduction, or permanent discontinuation based on severity and duration of LFT abnormality [see Dosage and Administration (2.3) and Use in Specific Populations (8.6)]. 5.4 Cardiomyopathy Cardiomyopathy including cardiac failure, congestive heart failure, ejection fraction decreased, diastolic dysfunction, or right ventricular dysfunction can occur with YONDELIS. In Trial 1, patients with a history of New York Heart Association Class II to IV heart failure or abnormal left ventricular ejection fraction (LVEF) at baseline were ineligible. In Trial 1, cardiomyopathy occurred in 23 patients (6%) receiving YONDELIS and in four patients (2.3%) receiving dacarbazine. Grade 3 or 4 cardiomyopathy occurred in 15 patients (4%) receiving YONDELIS and 2 patients (1.2%) receiving dacarbazine; cardiomyopathy leading to death occurred in 1 patient (0.3%) receiving YONDELIS and in none of the patients receiving dacarbazine. The median time to development of Grade 3 or 4 cardiomyopathy in patients receiving YONDELIS was 5.3 months (range: 26 days to 15.3 months). Assess LVEF by echocardiogram or multigated acquisition (MUGA) scan before initiation of YONDELIS and at 2- to 3-month intervals thereafter until YONDELIS is discontinued. Withhold YONDELIS for LVEF below lower limit of normal. Permanently discontinue YONDELIS for symptomatic cardiomyopathy or persistent left ventricular dysfunction that does not recover to lower limit of normal within 3 weeks [see Dosage and Administration (2.3)]. 5.5 Extravasation Resulting in Tissue Necrosis Extravasation of YONDELIS, resulting in tissue necrosis requiring debridement, can occur. Evidence of tissue necrosis can occur more than 1 week after the extravasation. There is no specific antidote for extravasation of YONDELIS. Administer YONDELIS through a central venous line [see Dosage and Administration (2.5)]. 5.6 Embryofetal Toxicity Based on its mechanism of action, YONDELIS can cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential to use effective contraception during therapy and for at least 2 months after the last dose of YONDELIS. Advise males with female partners of reproductive potential to use effective contraception during therapy and for at least 5 months after the last dose of YONDELIS [see Use in Specific Populations (8.1, 8.3)]. 6 ADVERSE REACTIONS The following adverse reactions are discussed in more detail in other sections of the labeling: Anaphylaxis [see Contraindications (4)] Neutropenic Sepsis [see Warnings and Precautions (5.1)] Rhabdomyolysis [see Warnings and Precautions (5.2)] Hepatotoxicity [see Warnings and Precautions (5.3)] Cardiomyopathy [see Warnings and Precautions (5.4)] Extravasation Resulting in Tissue Necrosis [see Warnings and Precautions (5.5)] 6.1 Adverse Reactions in Clinical Trials Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The data described below reflect exposure to YONDELIS in 755 patients with soft tissue sarcoma including 197 (26%) patients exposed to YONDELIS for greater than or equal to 6 months and 57 (8%) patients exposed to YONDELIS for greater than or equal to 1 year. The safety of YONDELIS was evaluated in six open-label, single-arm trials, in which 377 patients received YONDELIS and one open-label, randomized, active-controlled clinical trial in which 378 patients received YONDELIS (Trial 1). All patients received YONDELIS at the recommended dosing regimen of 1.5 mg/m2 administered as an intravenous infusion over 24 hours once every 3 weeks (q3wk, 24-h). The median age was 54 years (range: 18 to 81 years), 63% were female, and all patients had metastatic soft tissue sarcoma. Tables 3 and 4 present selected adverse reactions and laboratory abnormalities, respectively, observed in Trial 1, an open-label, randomized (2:1), active-controlled trial in which 550 patients with previously treated leiomyosarcoma or liposarcoma (dedifferentiated, myxoid round cell, or pleomorphic) received YONDELIS 1.5 mg/m2 intravenous infusion over 24 hours once every 3 weeks (n=378) or dacarbazine 1000 mg/m2 intravenous infusion over 20 to 120 minutes once every 3 weeks (n=172) [see Clinical Studies (14)]. All patients treated with YONDELIS were required to receive dexamethasone 20 mg intravenous injection 30 minutes prior to start of the YONDELIS infusion. In Trial 1, patients had been previously treated with an anthracycline- and ifosfamide-containing regimen or with an anthracycline-containing regimen and one additional cytotoxic chemotherapy regimen. The trial excluded patients with known central nervous system metastasis, elevated serum bilirubin or significant chronic liver disease, such as cirrhosis or active hepatitis, and history of myocardial infarction within 6 months, history of New York Heart Association Class II to IV heart failure, or abnormal left ventricular ejection fraction at baseline. The median age of patients in Trial 1 was 57 years (range: 17 to 81 years), with 69% female, 77% White, 12% Black or African American, 4% Asian, and <1% American Indian or Alaska Native. The median duration of exposure to trabectedin was 13 weeks (range: 1 to 127 weeks) with 30% of patients exposed to YONDELIS for greater than 6 months and 7% of patients exposed to YONDELIS for greater than 1 year. In Trial 1, adverse reactions resulting in permanent discontinuation of YONDELIS occurred in 26% (98/378) of patients; the most common were increased liver tests (defined as ALT, AST, alkaline phosphatase, bilirubin) (5.6%), thrombocytopenia (3.4%), fatigue (1.6%), increased creatine phosphokinase (1.1%), and decreased ejection fraction (1.1%). Adverse reactions that led to dose reductions occurred in 42% (158/378) of patients treated with YONDELIS; the most common were increased liver tests (24%), neutropenia (including febrile neutropenia) (8%), thrombocytopenia (4.2%), fatigue (3.7%), increased creatine phosphokinase (2.4%), nausea (1.1%), and vomiting (1.1%). Adverse reactions led to dose interruptions in 52% (198/378) of patients treated with YONDELIS; the most common were neutropenia (31%), thrombocytopenia (15%), increased liver tests (6%), fatigue (2.9%), anemia (2.6%), increased creatinine (1.1%), and nausea (1.1%). The most common adverse reactions (≥20%) were nausea, fatigue, vomiting, constipation, decreased appetite, diarrhea, peripheral edema, dyspnea, and headache. The most common laboratory abnormalities (≥20%) were increases in AST or ALT, increased alkaline phosphatase, hypoalbuminemia, increased creatinine, increased creatine phosphokinase, anemia, neutropenia, and thrombocytopenia. Table 3: Selected Adverse Reactions* Occurring in ≥10% of Patients Receiving YONDELIS and at a Higher Incidence than in the Control Arm - Trial 1
Toxicity grade is based on NCI common toxicity criteria, version 4.0. Fatigue is a composite of the following adverse event terms: fatigue, asthenia, and malaise. Other clinically important adverse reactions observed in <10% of patients (N=755) with soft tissue sarcoma receiving YONDELIS were: Nervous system disorders: peripheral neuropathy, paresthesia, hypoesthesia. Respiratory, thoracic, and mediastinal disorders: pulmonary embolism. Table 4: Incidence of Selected Treatment-Emergent Laboratory Abnormalities* - Trial 1
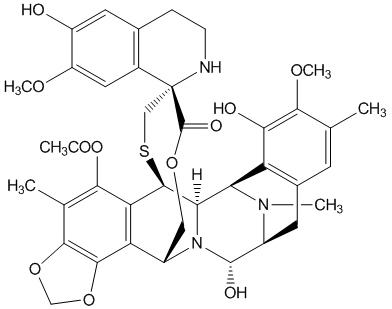
7 DRUG INTERACTIONS 7.1 Effect of Cytochrome CYP3A Inhibitors Coadministration of YONDELIS with ketoconazole, a strong CYP3A inhibitor, increased systemic exposure of trabectedin by 66%. Avoid using strong CYP3A inhibitors (e.g., oral ketoconazole, itraconazole, posaconazole, voriconazole, clarithromycin, telithromycin, indinavir, lopinavir, ritonavir, boceprevir, nelfinavir, saquinavir, telaprevir, nefazodone, conivaptan) in patients taking YONDELIS. Avoid taking grapefruit or grapefruit juice during YONDELIS treatment. If a strong CYP3A inhibitor for short-term use (i.e., less than 14 days) must be used, administer the strong CYP3A inhibitor 1 week after the YONDELIS infusion, and discontinue it the day prior to the next YONDELIS infusion [see Clinical Pharmacology (12.3)]. 7.2 Effect of Cytochrome CYP3A Inducers Coadministration of YONDELIS with rifampin, a strong CYP3A inducer, decreased systemic exposure of trabectedin by 31%. Avoid using strong CYP3A inducers (e.g., rifampin, phenobarbital, St. John's wort) in patients taking YONDELIS [see Clinical Pharmacology (12.3)]. 8 USE IN SPECIFIC POPULATIONS 8.1 Pregnancy Risk Summary Based on its mechanism of action, trabectedin can cause fetal harm when administered during pregnancy [see Clinical Pharmacology (12.1)]. There are no available data with the use of YONDELIS during pregnancy. Animal reproductive and developmental studies at relevant doses have not been conducted with trabectedin; however, placental transfer of trabectedin was demonstrated in pregnant rats. Advise pregnant woman of the potential risk to a fetus. The background risk of major birth defects and miscarriage for the indicated population are unknown; however, the background risk in the U.S. general population of major birth defects is 2 to 4% and of miscarriage is 15 to 20% of clinically recognized pregnancies. 8.2 Lactation Risk Summary There are no data on the presence of trabectedin in human milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for serious adverse reactions from YONDELIS in breastfed infants, advise a nursing woman to discontinue nursing during treatment with YONDELIS. 8.3 Females and Males of Reproductive Potential Contraception Females Advise female patients of reproductive potential to use effective contraception during and for 2 months after the last dose of YONDELIS [see Use in Specific Populations (8.1)]. Males YONDELIS may damage spermatozoa, resulting in possible genetic and fetal abnormalities. Advise males with a female sexual partner of reproductive potential to use effective contraception during and for 5 months after the last dose of YONDELIS [see Nonclinical Toxicology (13.1)]. Infertility YONDELIS may result in decreased fertility in males and females [see Nonclinical Toxicology (13.1)]. 8.4 Pediatric Use Safety and effectiveness in pediatric patients have not been established. 8.5 Geriatric Use Clinical studies of YONDELIS did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. 8.6 Hepatic Impairment The mean trabectedin exposure was (97%) higher in patients with moderate (bilirubin levels 1.5 to 3.0 times the upper limit of normal, and AST and ALT less than 8 times the upper limit of normal) hepatic impairment compared to patients with normal (total bilirubin ≤ the upper limit of normal, and AST and ALT ≤ the upper limit of normal) liver function. Reduce YONDELIS dose in patients with moderate hepatic impairment [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)]. Do not administer YONDELIS to patients with severe hepatic impairment (bilirubin levels above 3 times to 10 times the upper limit of normal, and any AST and ALT) [see Warnings and Precautions (5.3)]. 8.7 Renal Impairment No dose adjustment is recommended in patients with mild [creatinine clearance (CLcr) 60–89 mL/min] or moderate (CLcr of 30–59 mL/min) renal impairment. The pharmacokinetics of trabectedin has not been evaluated in patients with severe renal impairment (CLcr <30 mL/min) or end stage renal disease [see Clinical Pharmacology (12.3)]. 10 OVERDOSAGE There is no specific antidote for YONDELIS. Hemodialysis is not expected to enhance the elimination of YONDELIS because trabectedin is highly bound to plasma proteins (97%) and not significantly renally excreted. 11 DESCRIPTION Trabectedin is an alkylating agent with the chemical name (1'R,6R,6aR,7R,13S,14S,16R)-5-(acetyloxy)-3',4',6,6a,7,13,14,16-octahydro-6',8,14-trihydroxy-7',9-dimethoxy-4,10,23-trimethyl-spiro[6,16-(epithiopropanoxymethano)-7,13-imino-12H-1,3-dioxolo[7,8]isoquino[3,2-b][3]benzazocine-20,1'(2'H)-isoquinolin]-19-one. The molecular formula is C39H43N3O11S. The molecular weight is 761.84 daltons. The chemical structure is shown below:
Trabectedin is hydrophobic and has a low solubility in water.
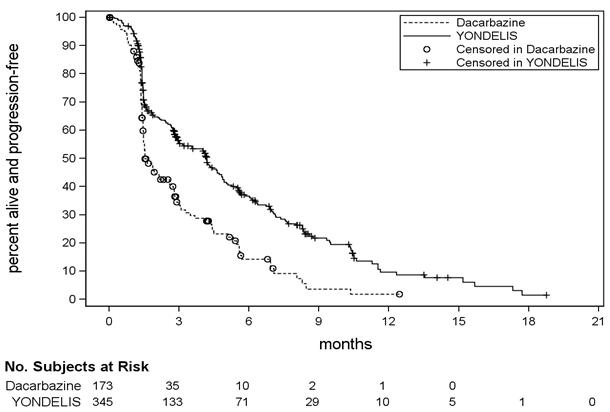
Unstratified log rank test. Based on 384 patients randomized to YONDELIS arm and 193 patients randomized to dacarbazine. Fisher's exact CI. Figure 1: Kaplan-Meier Curves of Progression-Free Survival in Trial 1
15 REFERENCES | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||