|
新型肺动脉高压口服药Uptravi(Selexipag Tablets)获美国FDA批准上市
2015年12月21日,美国食品和药品监管局(FDA)批准Uptravi (selexipag)片治疗成年 with 肺动脉高压(PAH),a慢性,逐步的,和使衰弱罕见的肺部疾病可导致死亡需要移植。
FDA的药品评价和研究中心中药品评价I部主任Ellis Unger,M.D.说:“Uptravi为有肺动脉高压患者提供一个另外治疗选择为,” “FDA支持为罕见病继续努力提供新治疗选择。”
PAH是在连接心脏至肺动脉发生高血压,它至心脏的右侧比正常更困难工作,可能导致远动能力受限制和气短,在其他更严重的并发症.
Uptravi属于一类药物称为口服IP前列环素受体激动剂。药物作用通过在血管壁放松肌肉扩张(开放)的血管和减低在血管中升高的压力供血至肺。
在一项1,156例有PAH参加者长期临床试验确定的安全性和疗效。与安慰剂比较,Uptravi被显示在减低为PAH住院有效和减低疾病进展的风险。在该试验参加者暴露于Uptravi中位时间1.4年。
在试验中用Uptravi患者观察到常见副作用包括头痛,腹泻,下颚痛,恶心,肌痛,呕吐,肢体痛,和脸红。
Uptravi被授予孤儿药物指定。孤儿药物指定提供激励例如税收减免,用户费用减免,和专有权资格帮助和鼓励对罕见病药物的开发。
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use UPTRAVI® safely and effectively. See full prescribing information for UPTRAVI®.
UPTRAVI® (selexipag) tablets, for oral use
Initial U.S. Approval: 2015
INDICATIONS AND USAGE
UPTRAVI® is a prostacyclin receptor agonist indicated for the treatment of pulmonary arterial hypertension (PAH, WHO Group I) to delay disease progression and reduce the risk of hospitalization for PAH. (1.1)
DOSAGE AND ADMINISTRATION
Starting dose: 200 mcg twice daily. (2.1)
Increase the dose by 200 mcg twice daily at weekly intervals to the highest tolerated dose up to 1600 mcg twice daily. (2.1)
Maintenance dose is determined by tolerability. (2.1)
Moderate hepatic impairment: Starting dose 200 mcg once daily, increase the dose by 200 mcg once daily at weekly intervals to the highest tolerated dose up to 1600 mcg. (2.3)
DOSAGE FORMS AND STRENGTHS
Tablets: 200 mcg, 400 mcg, 600 mcg, 800 mcg, 1000 mcg, 1200 mcg, 1400 mcg, 1600 mcg. (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
Pulmonary edema in patients with pulmonary veno-occlusive disease. If confirmed, discontinue treatment. (5.1)
ADVERSE REACTIONS
Adverse reactions occurring more frequently (≥5%) on UPTRAVI compared to placebo are headache, diarrhea, jaw pain, nausea, myalgia, vomiting, pain in extremity, and flushing. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Actelion at 1-866-228-3546 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Strong CYP2C8 inhibitors: increased exposure to selexipag and its active metabolite. Avoid concomitant use. (7.1, 12.3)
USE IN SPECIFIC POPULATIONS
Nursing mothers: discontinue UPTRAVI or breastfeeding. (8.2)
Severe hepatic impairment: Avoid use. (8.6)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2015
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Pulmonary Arterial Hypertension
UPTRAVI is indicated for the treatment of pulmonary arterial hypertension (PAH, WHO Group I) to delay disease progression and reduce the risk of hospitalization for PAH.
Effectiveness was established in a long-term study in PAH patients with WHO Functional Class II-III symptoms.
Patients had idiopathic and heritable PAH (58%), PAH associated with connective tissue disease (29%), PAH associated with congenital heart disease with repaired shunts (10%) [see Clinical Studies (14.1)].
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended starting dose of UPTRAVI is 200 micrograms (mcg) given twice daily. Tolerability may be improved when taken with food [see Clinical Pharmacology (12.3)].
Increase the dose in increments of 200 mcg twice daily, usually at weekly intervals, to the highest tolerated dose up to 1600 mcg twice daily. If a patient reaches a dose that cannot be tolerated, the dose should be reduced to the previous tolerated dose.
Do not split, crush, or chew tablets.
2.2 Interruptions and Discontinuations
If a dose of medication is missed, patients should take a missed dose as soon as possible unless the next dose is within the next 6 hours.
If treatment is missed for 3 days or more, restart UPTRAVI at a lower dose and then retitrate.
2.3 Dosage Adjustment in Patients with Hepatic Impairment
No dose adjustment of UPTRAVI is necessary for patients with mild hepatic impairment (Child-Pugh class A).
For patients with moderate hepatic impairment (Child-Pugh class B), the starting dose of UPTRAVI is 200 mcg once daily. Increase in increments of 200 mcg once daily at weekly intervals, as tolerated [see Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
Avoid use of UPTRAVI in patients with severe hepatic impairment (Child-Pugh class C).
3 DOSAGE FORMS AND STRENGTHS
UPTRAVI is available in the following strengths:
– 200 mcg [Light yellow tablet debossed with 2]
– 400 mcg [Red tablet debossed with 4]
– 600 mcg [Light violet tablet debossed with 6]
– 800 mcg [Green tablet debossed with 8]
– 1000 mcg [Orange tablet debossed with 10]
– 1200 mcg [Dark violet tablet debossed with 12]
– 1400 mcg [Dark yellow tablet debossed with 14]
– 1600 mcg [Brown tablet debossed with 16]
4 CONTRAINDICATIONS
None.
5 WARNINGS AND PRECAUTIONS
5.1 Pulmonary Veno-Occlusive Disease (PVOD)
Should signs of pulmonary edema occur, consider the possibility of associated PVOD. If confirmed, discontinue UPTRAVI.
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of UPTRAVI has been evaluated in a long-term, placebo-controlled study enrolling 1156 patients with symptomatic PAH (GRIPHON study) [see Clinical Studies (14)]. The exposure to UPTRAVI in this trial was up to 4.2 years with median duration of exposure of 1.4 years.
Table1 presents adverse reactions more frequent on UPTRAVI than on placebo by ≥3%.
Table 1 Adverse Reactions
| Adverse Reaction |
UPTRAVI
N=575
|
Placebo
N=577
|
| Headache |
65% |
32% |
| Diarrhea |
42% |
18% |
| Jaw pain |
26% |
6% |
| Nausea |
33% |
18% |
| Myalgia |
16% |
6% |
| Vomiting |
18% |
9% |
| Pain in Extremity |
17% |
8% |
| Flushing |
12% |
5% |
| Arthralgia |
11% |
8% |
| Anemia |
8% |
5% |
| Decreased appetite |
6% |
3% |
| Rash |
11% |
8% | These adverse reactions are more frequent during the dose titration phase.
Hyperthyroidism was observed in 1% (n=8) of patients on UPTRAVI and in none of the patients on placebo.
Laboratory Test Abnormalities
Hemoglobin
In a Phase 3 placebo-controlled study in patients with PAH, mean absolute changes in hemoglobin at regular visits compared to baseline ranged from −0.34 to −0.02 g/dL in the selexipag group compared to −0.05 to 0.25 g/dL in the placebo group. A decrease in hemoglobin concentration to below 10 g/dL was reported in 8.6% of patients treated with selexipag and 5.0% of placebo-treated patients.
Thyroid function tests
In a Phase 3 placebo-controlled study in patients with PAH, a reduction (up to −0.3 MU/L from a baseline median of 2.5 MU/L) in median thyroid-stimulating hormone (TSH) was observed at most visits in the selexipag group. In the placebo group, little change in median values was apparent. There were no mean changes in triiodothyronine or thyroxine in either group.
7 DRUG INTERACTIONS
7.1 Strong CYP2C8 Inhibitors
Concomitant administration with strong inhibitors of CYP2C8 may result in a significant increase in exposure to selexipag and its active metabolite. Avoid concomitant administration of UPTRAVI with strong inhibitors of CYP2C8 (e.g., gemfibrozil) [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies with UPTRAVI in pregnant women. Animal reproduction studies performed with selexipag showed no clinically relevant effects on embryofetal development and survival. A slight reduction in maternal as well as in fetal body weight was observed when pregnant rats were administered selexipag during organogenesis at a dose producing an exposure approximately 47 times that in humans at the maximum recommended human dose. No adverse developmental outcomes were observed with oral administration of selexipag to pregnant rabbits during organogenesis at exposures up to 50 times the human exposure at the maximum recommended human dose.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Pregnant rats were treated with selexipag using oral doses of 2, 6, and 20 mg/kg/day (up to 47 times the exposure at the maximum recommended human dose of 1600 mcg twice daily on an area under the curve [AUC] basis) during the period of organogenesis (gestation days 7 to 17). Selexipag did not cause adverse developmental effects to the fetus in this study. A slight reduction in fetal body weight was observed in parallel with a slight reduction in maternal body weight at the high dose.
Pregnant rabbits were treated with selexipag using oral doses of 3, 10, and 30 mg/kg (up to 50 times the exposure to the active metabolite at the maximum recommended human dose of 1600 mcg twice daily on an AUC basis) during the period of organogenesis (gestation days 6 to 18). Selexipag did not cause adverse developmental effects to the fetus in this study.
8.2 Lactation
It is not known if UPTRAVI is present in human milk. Selexipag or its metabolites were present in the milk of rats. Because many drugs are present in the human milk and because of the potential for serious adverse reactions in nursing infants, discontinue nursing or discontinue UPTRAVI.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
8.5 Geriatric Use
Of the 1368 subjects in clinical studies of UPTRAVI 248 subjects were 65 years of age and older, while 19 were 75 and older. No overall differences were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity cannot be ruled out.
8.6 Patients with Hepatic Impairment
No adjustment to the dosing regimen is needed in patients with mild hepatic impairment (Child-Pugh class A).
A once-daily regimen is recommended in patients with moderate hepatic impairment (Child-Pugh class B) due to the increased exposure to selexipag and its active metabolite. There is no experience with UPTRAVI in patients with severe hepatic impairment (Child-Pugh class C). Avoid use of UPTRAVI in patients with severe hepatic impairment [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
8.7 Patients with Renal Impairment
No adjustment to the dosing regimen is needed in patients with estimated glomerular filtration rate > 15 mL/min/1.73 m2.
There is no clinical experience with UPTRAVI in patients undergoing dialysis or in patients with glomerular filtration rates < 15 mL/min/1.73 m2 [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
Isolated cases of overdose up to 3200 mcg were reported. Mild, transient nausea was the only reported consequence. In the event of overdose, supportive measures must be taken as required. Dialysis is unlikely to be effective because selexipag and its active metabolite are highly protein-bound.
11 DESCRIPTION
UPTRAVI (selexipag) is a selective non-prostanoid IP prostacyclin receptor agonist. The chemical name of selexipag is 2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl) acetamide. It has a molecular formula of C26H32N4O4S and a molecular weight of 496.62. Selexipag has the following structural formula:

Selexipag is a pale yellow crystalline powder that is practically insoluble in water. In the solid state selexipag is very stable, is not hygroscopic, and is not light sensitive.
Depending on the dose strength, each round film-coated tablet for oral administration contains 200, 400, 600, 800, 1000, 1200, 1400, or 1600 mcg of selexipag. The tablets include the following inactive ingredients: D-mannitol, corn starch, low substituted hydroxypropylcellulose, hydroxypropylcellulose, and magnesium stearate. The tablets are film coated with a coating material containing hypromellose, propylene glycol, titanium dioxide, carnauba wax along with mixtures of iron oxide red, iron oxide yellow or iron oxide black.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Selexipag is an oral prostacyclin receptor (IP receptor) agonist that is structurally distinct from prostacyclin. Selexipag is hydrolyzed by carboxylesterase 1 to yield its active metabolite, which is approximately 37-fold as potent as selexipag. Selexipag and the active metabolite are selective for the IP receptor versus other prostanoid receptors (EP1-4, DP, FP and TP).
12.2 Pharmacodynamics
Cardiac electrophysiology:
At the maximum tolerated dose of 1600 mcg twice daily, selexipag does not prolong the QT interval to any clinically relevant extent.
Platelet aggregation:
Both selexipag and its active metabolite caused concentration-dependent inhibition of platelet aggregation in vitro with an IC50 of 5.5 µM and 0.21 µM, respectively. However, at clinically relevant concentrations, there was no effect on platelet aggregation test parameters as seen following multiple-dose administrations of selexipag in healthy subjects from 400 mcg up to 1800 mcg twice daily.
Pulmonary hemodynamics:
A Phase 2 clinical study assessed hemodynamic variables after 17 weeks of treatment in patients with PAH WHO Functional Class II–III and concomitantly receiving endothelin receptor antagonists (ERAs) and/or phosphodiesterase type 5 (PDE-5) inhibitors. Patients titrating selexipag to an individually tolerated dose (200 mcg twice daily increments up to 800 mcg twice daily) (N=33) achieved a statistically-significant mean reduction in pulmonary vascular resistance of 30.3% (95% confidence interval [CI] −44.7%, −12.2%) and an increase in cardiac index (median treatment effect) of 0.41 L/min/m2 (95% CI 0.10, 0.71) compared to placebo (N=10).
Drug interaction:
In a study in healthy subjects, selexipag (400 mcg twice a day) did not influence the pharmacodynamic effect of warfarin on the international normalized ratio.
12.3 Pharmacokinetics
The pharmacokinetics of selexipag and its active metabolite have been studied primarily in healthy subjects. The pharmacokinetics of selexipag and the active metabolite, after both single- and multiple-dose administration, were dose-proportional up to a single dose of 800 mcg and multiple doses of up to 1800 mcg twice daily. No accumulation in plasma, either of parent compound or active metabolite, occurred after multiple-dose administration.
In healthy subjects, inter-subject variability in exposure (area under the curve over a dosing interval, AUC) at steady-state was 43% and 39% for selexipag and the active metabolite, respectively. Intra-subject variability in exposure was 24% and 19% for selexipag and the active metabolite, respectively.
Exposures to selexipag and the active metabolite at steady-state in PAH patients and healthy subjects were similar. The pharmacokinetics of selexipag and the active metabolite in PAH patients were not influenced by the severity of the disease and did not change with time.
Both in healthy subjects and PAH patients, exposure at steady-state to the active metabolite is approximately 3- to 4-fold that of selexipag.
Absorption
Upon oral administration, maximum observed plasma concentrations of selexipag and its active metabolite after oral administration are reached within about 1–3 hours and 3–4 hours, respectively.
In the presence of food, the absorption of selexipag was prolonged resulting in a delayed time to peak concentration (Tmax) and ~30% lower peak plasma concentration (Cmax). The exposure to selexipag and the active metabolite (AUC) did not significantly change in the presence of food.
Distribution
Selexipag and its active metabolite are highly bound to plasma proteins (approximately 99% in total and to the same extent to albumin and alpha1-acid glycoprotein).
Metabolism
Selexipag undergoes enzymatic hydrolysis of the acylsulfonamide by hepatic carboxylesterase 1, to yield the active metabolite. Oxidative metabolism catalyzed by CYP3A4 and CYP2C8 leads to the formation of hydroxylated and dealkylated products. UGT1A3 and UGT2B7 are involved in the glucuronidation of the active metabolite. Except for the active metabolite, none of the circulating metabolites in human plasma exceeds 3% of the total drug-related material.
Elimination
Elimination of selexipag is predominately via metabolism with a mean terminal half-life of 0.8-2.5 hours. The active metabolite has a terminal half-life of 6.2-13.5 hours. The apparent oral clearance of selexipag is on average 35 L/hour.
Excretion
In a study in healthy subjects with radiolabeled selexipag, approximately 93% of radioactive drug material was eliminated in feces and only 12% in urine. Neither selexipag nor its active metabolite were found in urine.
Specific Populations:
No clinically relevant effects of sex, race, age or body weight on the pharmacokinetics of selexipag and its active metabolite have been observed in healthy subjects or PAH patients.
Age:
The pharmacokinetic variables (Cmax and AUC) were similar in adult and elderly subjects up to 75 years of age. There was no effect of age on the pharmacokinetics of selexipag and the active metabolite in PAH patients.
Hepatic Impairment:
In subjects with mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment, exposure to selexipag was 2- and 4-fold that seen in healthy subjects. Exposure to the active metabolite of selexipag remained almost unchanged in subjects with mild hepatic impairment and was doubled in subjects with moderate hepatic impairment. [see Use in Specific Populations (8.6)].
Baed on pharmacokinetic modeling of data from a study in subjects with hepatic impairment, the exposure to the active metabolite at steady state in subjects with moderate hepatic impairment (Child-Pugh class B) after a once daily regimen is expected to be similar to that in healthy subjects receiving a twice daily regimen. The exposure to selexipag at steady state in these patients during a once daily regimen is predicted to be approximately 2-fold that seen in healthy subjects receiving a twice-daily regimen.
Renal Impairment:
A 40-70% increase in exposure (maximum plasma concentration and area under the plasma concentration-time curve) to selexipag and its active metabolite was observed in subjects with severe renal impairment (estimated glomerular filtration rate ≥ 15 mL/min/1.73 m2 and < 30 mL/min/1.73 m2) [see Use in Specific Populations (8.7)].
Drug Interaction Studies:
In vitro studies
Selexipag is hydrolyzed to its active metabolite by hepatic carboxylesterase 1. Selexipag and its active metabolite both undergo oxidative metabolism by CYP2C8 and CYP3A4. The glucuronidation of the active metabolite is catalyzed by UGT1A3 and UGT2B7. Selexipag and its active metabolite are substrates of OATP1B1 and OATP1B3. Selexipag is a substrate of P-gp, and the active metabolite is a substrate of the transporter of breast cancer resistance protein (BCRP).
Selexipag and its active metabolite do not inhibit or induce hepatic cytochrome P450 enzymes at clinically relevant concentrations. Selexipag and its active metabolite do not inhibit hepatic or renal transport proteins.
The effect of strong inhibitors of CYP2C8 (such as gemfibrozil) on the exposure to selexipag or its active metabolite has not been studied. Concomitant administration with strong inhibitors of CYP2C8 may result in a significant increase in exposure to selexipag and its active metabolite [see Drug Interactions (7.1)].
The results on in vivo drug interaction studies are presented in Figure 1.
Figure 1 Effect of Other Drugs on UPTRAVI and its Active Metabolite (A) and Effect of UPTRAVI on Warfarin (B)

ERA and PDE-5 inhibitor data from GRIPHON.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: In the 2-year carcinogenicity studies, chronic oral administration of selexipag revealed no evidence of carcinogenic potential in rats at 100 mg/kg/day and mice at 500 mg/kg/day. The exposures were more than 25-fold human exposure.
Mutagenesis: Selexipag and the active metabolite are not genotoxic on the basis of the overall evidence of conducted genotoxicity studies.
Fertility: The no effect dose for effects on fertility was 60 mg/kg/day in a study in which rats were administered selexipag orally. This dose corresponded to an exposure of 175-times (active metabolite) the human therapeutic exposure.
14 CLINICAL STUDIES
14.1 Pulmonary Arterial Hypertension
The effect of selexipag on progression of PAH was demonstrated in a multi-center, double-blind, placebo-controlled, parallel group, event-driven study (GRIPHON) in 1156 patients with symptomatic (WHO Functional Class I [0.8%], II [46%], III [53%], and IV [1%] ) PAH. Patients were randomized to either placebo (N = 582), or UPTRAVI (N = 574). The dose was increased in weekly intervals by increments of 200 mcg twice a day to the highest tolerated dose up to 1600 mcg twice a day.
The primary study endpoint was the time to first occurrence up to end-of-treatment of: a) death, b) hospitalization for PAH, c) PAH worsening resulting in need for lung transplantation, or balloon atrial septostomy, d) initiation of parenteral prostanoid therapy or chronic oxygen therapy, or e) other disease progression based on a 15% decrease from baseline in 6MWD plus worsening of Functional Class or need for additional PAH-specific therapy.
The mean age was 48 years, the majority of patients were white (65%) and female (80%). Nearly all patients were in WHO Functional Class II and III at baseline.
Idiopathic or heritable PAH was the most common etiology in the study population (58%) followed by PAH associated with connective tissue disease (29%), PAH associated with congenital heart disease with repaired shunts (10%), drugs and toxins (2%), and HIV (1%).
At baseline, the majority of enrolled patients (80%) were being treated with a stable dose of an endothelin receptor antagonist (15%), a PDE-5 inhibitor (32%), or both (33%).
Patients on selexipag achieved doses within the following groups: 200 - 400 mcg (23%), 600 - 1000 mcg (31%) and 1200 - 1600 mcg (43%).
Treatment with UPTRAVI resulted in a 40% reduction (99% CI: 22 to 54%; two-sided log-rank p-value < 0.0001) of the occurrence of primary endpoint events compared to placebo (Table 2; Figure 2). The beneficial effect of UPTRAVI was primarily attributable to a reduction in hospitalization for PAH and a reduction in other disease progression events (Table 2). The observed benefit of UPTRAVI was similar regardless of the dose achieved when patients are titrated to their highest tolerated dose [see Dosage and Administration (2.1)].
Figure 2 Kaplan-Meier Estimates of the First Morbidity-Mortality Event in GRIPHON
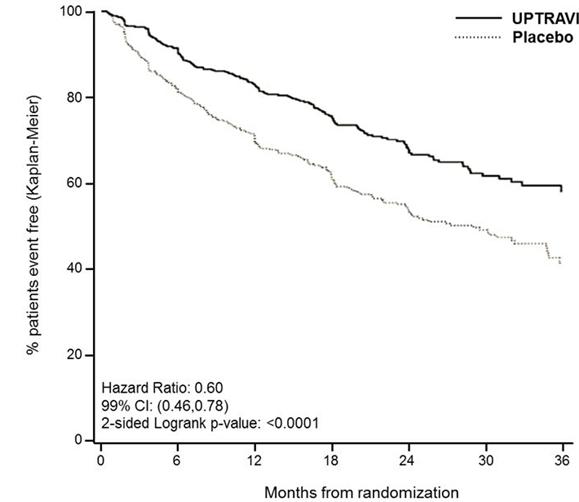

Table 2 Primary Endpoints and Related Components in GRIPHON
UPTRAVI
N=574 |
Placebo
N=582 |
Hazard Ratio
(99% CI) |
p-value |
|
n |
% |
n |
% |
|
|
| Primary endpoint events up to the end of treatment |
| All primary endpoint events |
155 |
27.0 |
242 |
41.6 |
0.60 [0.46,0.78] |
<0.0001 |
| As first event: |
|
|
|
|
|
|
|
|
78 |
13.6 |
109 |
18.7 |
|
|
- Other disease Progression
(Decrease in 6MWD plus
worsening functional class or
need for other therapy) |
38 |
6.6 |
100 |
17.2 |
|
|
|
|
28 |
4.9 |
18 |
3.1 |
|
|
- Parenteral prostanoid or
chronic oxygen therapy |
10 |
1.7 |
13 |
2.2 |
|
|
- PAH worsening resulting in
need for lung transplantation
or balloon atrial septostomy |
1 |
0.2 |
2 |
0.3 | It is not known if the excess number of deaths in the selexipag group is drug-related because there were so few deaths and the imbalance was not observed until 18 months into GRIPHON.
Figures 3A, B and C show time to first event analyses for primary endpoint components of hospitalization for PAH (A), other disease progression (B), and death (C)—all censored 7 days after any primary end point event (because many patients on placebo transitioned to open-label UPTRAVI at this point).
Figure 3 A Hospitalization for PAH as the First Endpoint in GRIPHON

Figure 3B Disease Progression as the First Endpoint in GRIPHON

Figure 3 C Death as the First Endpoint in GRIPHON

The treatment effect of UPTRAVI on time to first primary event was consistent irrespective of background PAH therapy (i.e., in combination with ERA, PDE5i, or both, or without background therapy) (Figure 4).
Figure 4 Subgroup Analyses of the Primary Endpoint in GRIPHON

Note: Race group “Other” is not displayed in analysis, as the population is less than 30. EU = Number of UPTRAVI patients with events, NU = Number of patients randomized to UPTRAVI, EP = Number of Placebo patients with events, NP = Number of patients randomized to Placebo, HR = Hazard Ratio, CI = Confidence Interval, the size of the squares represent the number of patients in the subgroup.
Note: The figure above presents effects in various subgroups all of which are baseline characteristics and all were pre-specified. The 99% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
6-Minute Walk Distance (6MWD)
Exercise capacity was evaluated as a secondary endpoint. Median absolute change from baseline to week 26 in 6MWD measured at trough (i.e. at approximately 12 hours post-dose) was +4 meters with UPTRAVI and -9 meters in the placebo group. This resulted in a placebo-corrected median treatment effect of 12 meters (99% CI: 1, 24 meters;two-sided p = 0.005).
16 HOW SUPPLIED/STORAGE AND HANDLING
UPTRAVI (selexipag) film-coated, round tablets are supplied in the following configurations:
Strength
(mcg) |
Color |
Debossing |
NDC-XXX
Bottle of 60 |
NDC-XXX
Bottle of 140 |
| 200 |
Light yellow |
2 |
66215-602-06 |
66215-602-14 |
| 400 |
Red |
4 |
66215-604-06 |
Not Applicable |
| 600 |
Light violet |
6 |
66215-606-06 |
Not Applicable |
| 800 |
Green |
8 |
66215-608-06 |
Not Applicable |
| 1000 |
Orange |
10 |
66215-610-06 |
Not Applicable |
| 1200 |
Dark violet |
12 |
66215-612-06 |
Not Applicable |
| 1400 |
Dark yellow |
14 |
66215-614-06 |
Not Applicable |
| 1600 |
Brown |
16 |
66215-616-06 |
Not Applicable | UPTRAVI is also supplied in a Titration Pack [NDC 66215-628-20] that includes a 140 count bottle of 200 mcg tablets and a 60 count bottle of 800 mcg tablets.
Store at 20°C to 25°C (68°F to 77°F). Excursions are permitted between 15°C and 30°C (59°F and 86°F). [See USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Package Insert).
http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=a7a23b87-f892-4e2c-8e2e-ebf841220f90 |
