|
英文药名:Tafinlar(dabrafenib capsules) 中文药名:达拉菲尼胶囊 生产厂家:GlaxoSmithKline
bSee Table 1 for recommended dose reductions of TAFINLAR and trametinib. cRefer to Full Prescribing Information for trametinib. 3 DOSAGE FORMS AND STRENGTHS 50 mg Capsules: Dark red capsule imprinted with ‘GS TEW’ and ‘50 mg’. 75 mg Capsules: Dark pink capsule imprinted with ‘GS LHF’ and ‘75 mg’. 4 CONTRAINDICATIONS None. 5 WARNINGS AND PRECAUTIONS Review the Full Prescribing Information for trametinib prior to initiation of TAFINLAR in combination with trametinib. The following serious adverse reactions of trametinib as a single agent, which may occur when TAFINLAR is used in combination with trametinib, are not described in the Full Prescribing Information for TAFINLAR: • Retinal vein occlusion • Interstitial lung disease 5.1 New Primary Malignancies New primary malignancies, cutaneous and non-cutaneous, can occur when TAFINLAR is administered as a single agent or when used in combination with trametinib. Cutaneous Malignancies: TAFINLAR results in an increased incidence of cutaneous squamous cell carcinoma, keratoacanthoma, and melanoma. TAFINLAR when used in combination with trametinib results in an increased incidence of basal cell carcinoma. In Trial 1, cutaneous squamous cell carcinomas and keratoacanthomas (cuSCC) occurred in 7% (14/187) of patients treated with TAFINLAR and in none of the patients treated with dacarbazine. Across clinical trials of TAFINLAR (N = 586), the incidence of cuSCC was 11%. The median time to first cuSCC was 9 weeks (range: 1 to 53 weeks). Of those patients who developed new cuSCC, approximately 33% developed one or more cuSCC with continued administration of TAFINLAR. The median time between diagnosis of the first cuSCC and the second cuSCC was 6 weeks. In Trial 1, the incidence of new primary malignant melanomas was 2% (3/187) for patients receiving TAFINLAR while no dacarbazine-treated patient was diagnosed with new primary malignant melanoma. In Trial 2, the incidence of basal cell carcinoma was increased in patients receiving TAFINLAR in combination with trametinib: 9% (5/55) of patients receiving TAFINLAR in combination with trametinib compared with 2% (1/53) of patients receiving TAFINLAR as a single agent. The range of time to diagnosis of basal cell carcinoma was 28 to 249 days in patients receiving TAFINLAR in combination with trametinib and was 197 days for the patient receiving TAFINLAR as a single agent. Cutaneous squamous cell carcinoma (SCC), including keratoacanthoma, occurred in 7% of patients receiving TAFINLAR in combination with trametinib and 19% of patients receiving TAFINLAR as a single agent. The range of time to diagnosis of cuSCC was 136 to197 days in the combination arm and was 9 to 197 days in the arm receiving TAFINLAR as a single agent. New primary melanoma occurred in 2% (1/53) of patients receiving TAFINLAR as a single agent and in none of the 55 patients receiving TAFINLAR in combination with trametinib. Perform dermatologic evaluations prior to initiation of TAFINLAR as a single agent or in combination with trametinib, every 2 months while on therapy, and for up to 6 months following discontinuation of TAFINLAR. No dose modifications of TAFINLAR or trametinib are required in patients who develop new primary cutaneous malignancies. Non-cutaneous Malignancies: Based on its mechanism of action, TAFINLAR may promote the growth and development of malignancies with activation of RAS through mutation or other mechanisms [see Warnings and Precautions (5.2)]. In patients receiving TAFINLAR in combination with trametinib four cases of non-cutaneous malignancies were identified: KRAS mutation-positive pancreatic adenocarcinoma (n = 1), recurrent NRAS mutation-positive colorectal carcinoma (n = 1), head and neck carcinoma (n = 1), and glioblastoma (n = 1). Monitor patients receiving the combination closely for signs or symptoms of non-cutaneous malignancies. Permanently discontinue TAFINLAR for RAS mutation-positive non-cutaneous malignancies. If used in combination with trametinib, no dose modification of trametinib is required for patients who develop non-cutaneous malignancies. 5.2 Tumor Promotion in BRAF Wild-Type Melanoma In vitro experiments have demonstrated paradoxical activation of MAP-kinase signaling and increased cell proliferation in BRAF wild-type cells which are exposed to BRAF inhibitors. Confirm evidence of BRAF V600E or V600K mutation status prior to initiation of TAFINLAR as a single agent or combination therapy [see Indications and Usage (1), Dosage and Administration (2.1)]. 5.3 Hemorrhage Hemorrhages, including major hemorrhages defined as symptomatic bleeding in a critical area or organ, can occur when TAFINLAR is used in combination with trametinib. In Trial 2, treatment with TAFINLAR in combination with trametinib resulted in an increased incidence and severity of any hemorrhagic event: 16% (9/55) of patients treated with TAFINLAR in combination with trametinib compared with 2% (1/53) of patients treated with TAFINLAR as a single agent. The major hemorrhagic events of intracranial or gastric hemorrhage occurred in 5% (3/55) of patients treated with TAFINLAR in combination with trametinib compared with none of the 53 patients treated with TAFINLAR as a single agent. Intracranial hemorrhage was fatal in 4% (2/55) of patients receiving TAFINLAR in combination with trametinib. Permanently discontinue TAFINLAR and trametinib for all Grade 4 hemorrhagic events and for any Grade 3 hemorrhagic events that do not improve. Withhold TAFINLAR for Grade 3 hemorrhagic events; if improved resume at a lower dose level. Withhold trametinib for up to 3 weeks for Grade 3 hemorrhagic events; if improved, resume at a lower dose level. 5.4 Venous Thromboembolism Venous thromboembolism can occur when TAFINLAR is used in combination with trametinib. In Trial 2, treatment with TAFINLAR in combination with trametinib resulted in an increased incidence of deep venous thrombosis (DVT) and pulmonary embolism (PE): 7% (4/55) of patients treated with TAFINLAR in combination with trametinib compared with none of the 53 patients treated with TAFINLAR as a single agent. Pulmonary embolism was fatal in 2% (1/55) of patients receiving TAFINLAR in combination with trametinib. Advise patients to immediately seek medical care if they develop symptoms of DVT or PE, such as shortness of breath, chest pain, or arm or leg swelling. Permanently discontinue TAFINLAR and trametinib for life-threatening PE. Withhold trametinib and continue TAFINLAR at the same dose for uncomplicated DVT or PE; if improved within 3 weeks, trametinib may be resumed at a lower dose level [see Dosage and Administration (2.3)]. 5.5 Cardiomyopathy Cardiomyopathy can occur when TAFINLAR is used in combination with trametinib and with trametinib as a single agent [refer to Full Prescribing Information for trametinib]. In Trial 2, cardiomyopathy occurred in 9% (5/55) of patients treated with TAFINLAR in combination with trametinib and in none of patients treated with TAFINLAR as a single agent. The median time to onset of cardiomyopathy in patients treated with TAFINLAR in combination with trametinib was 86 days (range: 27 to 253 days). Cardiomyopathy was identified within the first month of treatment with TAFINLAR in combination with trametinib in two of five patients. Development of cardiomyopathy resolved in all five patients following dose reduction (4/55) and/or dose interruption (1/55). Across clinical trials of TAFINLAR administered in combination with trametinib (N = 202), 8% of patients developed evidence of cardiomyopathy (decrease in LVEF below institutional lower limits of normal with an absolute decrease in LVEF ≥10% below baseline). Two percent demonstrated a decrease in LVEF below institutional lower limits of normal with an absolute decrease in LVEF of ≥20% below baseline. Assess LVEF by echocardiogram or multigated acquisition (MUGA) scan before initiation of TAFINLAR in combination with trametinib, one month after initiation, and then at 2- to 3-month intervals while on treatment with the combination. Withhold treatment with trametinib and continue TAFINLAR at the same dose if absolute LVEF value decreases by 10% from pretreatment values and is less than the lower limit of normal. For symptomatic cardiomyopathy or persistent, asymptomatic LV dysfunction that does not resolve within 4 weeks, permanently discontinue trametinib and withhold TAFINLAR. Resume TAFINLAR at the same dose level upon recovery of cardiac function [see Dosage and Administration (2.3)]. 5.6 Ocular Toxicities Retinal Pigment Epithelial Detachment (RPED): Retinal pigment epithelial detachments (RPED) can occur when TAFINLAR is used in combination with trametinib and with trametinib as a single agent [refer to Full Prescribing Information for trametinib]. Retinal detachments resulting from trametinib are often bilateral and multifocal, occurring in the macular region of the retina. In Trial 2, ophthalmologic examinations including retinal evaluation were performed pretreatment and at regular intervals during treatment. RPED occurred in 2% (1/55) of patients receiving TAFINLAR in combination with trametinib. Across clinical trials of TAFINLAR administered in combination with trametinib (N = 202), the incidence of RPED was 1% (2/202). Perform ophthalmological evaluation at any time a patient reports visual disturbances and compare with baseline, if available. If TAFINLAR is used in combination with trametinib, do not modify the dose of TAFINLAR. Withhold trametinib if RPED is diagnosed. If resolution of the RPED is documented on repeat ophthalmological evaluation within 3 weeks, resume trametinib at a lower dose level. Discontinue trametinib if no improvement after 3 weeks [see Dosage and Administration (2.3)]. Uveitis and Iritis: Uveitis and iritis can occur when TAFINLAR is administered as a single agent or when used in combination with trametinib. Uveitis (including iritis) occurred in 1% (6/586) of patients treated with TAFINLAR as a single agent and uveitis occurred in 1% (2/202) of patients treated with TAFINLAR in combination with trametinib. Symptomatic treatment employed in clinical trials included steroid and mydriatic ophthalmic drops. Monitor patients for visual signs and symptoms of uveitis (e.g., change in vision, photophobia, eye pain). If diagnosed, withhold TAFINLAR for up to 6 weeks until uveitis/iritis resolves to Grade 0-1. If TAFINLAR is used in combination with trametinib, do not modify the dose of trametinib. 5.7 Serious Febrile Reactions Serious febrile reactions and fever of any severity complicated by hypotension, rigors or chills, dehydration, or renal failure, can occur when TAFINLAR is administered as a single agent or when used in combination with trametinib. The incidence and severity of pyrexia are increased when TAFINLAR is used in combination with trametinib compared with TAFINLAR as a single agent [see Adverse Reactions (6.1)]. In Trial 1, the incidence of fever (serious and non-serious) was 28% in patients treated with TAFINLAR and 10% in patients treated with dacarbazine. In patients treated with TAFINLAR, the median time to initial onset of fever (any severity) was 11 days (range: 1 to 202 days) and the median duration of fever was 3 days (range: 1 to 129 days). Serious febrile reactions and fever of any severity complicated by hypotension, rigors or chills occurred in 3.7% (7/187) of patients treated with TAFINLAR and in none of the 59 patients treated with dacarbazine. In Trial 2, the incidence of fever (serious and non-serious) was 71% (39/55) in patients treated with TAFINLAR in combination with trametinib and 26% (14/53) in patients treated with TAFINLAR as a single agent. Serious febrile reactions and fever of any severity complicated by hypotension, rigors or chills occurred in 25% (14/55) of patients treated with TAFINLAR in combination with trametinib compared with 2% (1/53) of patients treated with TAFINLAR as a single agent. Fever was complicated with chills/rigors in 51% (28/55), dehydration in 9% (5/55), renal failure in 4% (2/55), and syncope in 4% (2/55) of patients in Trial 2. In patients treated with TAFINLAR in combination with trametinib, the median time to initial onset of fever was 30 days compared with 19 days in patients treated with TAFINLAR as a single agent; the median duration of fever was 6 days with the combination compared with 4 days with TAFINLAR as a single agent. Across clinical trials of TAFINLAR administered in combination with trametinib (N = 202), the incidence of pyrexia was 57% (116/202). Withhold TAFINLAR for fever of 101.3ºF or higher. Withhold trametinib for any fever higher than 104ºF. Withhold TAFINLAR, and trametinib if used in combination, for any serious febrile reaction or fever complicated by hypotension, rigors or chills, dehydration, or renal failure and evaluate for signs and symptoms of infection. Refer to Table 2 for recommended dose modifications for adverse reactions [see Dosage and Administration (2.3)]. Prophylaxis with antipyretics may be required when resuming TAFINLAR or trametinib. 5.8 Serious Skin Toxicity Serious skin toxicity can occur when TAFINLAR is used in combination with trametinib and with trametinib as a single agent [refer to Full Prescribing Information for trametinib]. In Trial 2, the incidence of any skin toxicity was similar for patients receiving TAFINLAR in combination with trametinib (65% [36/55]) compared with patients receiving TAFINLAR as a single agent (68% [36/53]). The median time to onset of skin toxicity in patients treated with TAFINLAR in combination with trametinib was 37 days (range: 1 to 225 days) and median time to resolution of skin toxicity was 33 days (range: 3 to 421 days). No patient required dose reduction or permanent discontinuation of TAFINLAR or trametinib for skin toxicity. Across clinical trials of TAFINLAR in combination with trametinib (N = 202), severe skin toxicity and secondary infections of the skin requiring hospitalization occurred in 2.5% (5/202) of patients treated with TAFINLAR in combination with trametinib. Withhold TAFINLAR, and trametinib if used in combination, for intolerable or severe skin toxicity. TAFINLAR and trametinib may be resumed at lower dose levels in patients with improvement or recovery from skin toxicity within 3 weeks [see Dosage and Administration (2.3)]. 5.9 Hyperglycemia Hyperglycemia can occur when TAFINLAR is administered as a single agent or when used in combination with trametinib. In Trial 1, 5 of 12 patients with a history of diabetes required more intensive hypoglycemic therapy while taking TAFINLAR. The incidence of Grade 3 hyperglycemia based on laboratory values was 6% (12/187) in patients treated with TAFINLAR compared with none of the dacarbazine-treated patients. In Trial 2, the incidence of Grade 3 hyperglycemia based on laboratory values was 5% (3/55) in patients treated with TAFINLAR in combination with trametinib compared with 2% (1/53) in patients treated with TAFINLAR as a single agent. Monitor serum glucose levels as clinically appropriate when TAFINLAR is administered as a single agent or when used in combination with trametinib in patients with pre-existing diabetes or hyperglycemia. Advise patients to report symptoms of severe hyperglycemia such as excessive thirst or any increase in the volume or frequency of urination. 5.10 Glucose-6-Phosphate Dehydrogenase Deficiency TAFINLAR, which contains a sulfonamide moiety, confers a potential risk of hemolytic anemia in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Closely observe patients with G6PD deficiency for signs of hemolytic anemia. 5.11 Embryofetal Toxicity Based on its mechanism of action, TAFINLAR can cause fetal harm when administered to a pregnant woman. Dabrafenib was teratogenic and embryotoxic in rats at doses three times greater than the human exposure at the recommended clinical dose. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations (8.1)]. Advise female patients of reproductive potential to use a highly effective non-hormonal method of contraception since TAFINLAR can render hormonal contraceptives ineffective, during treatment and for at least 2 weeks after treatment with TAFINLAR or for 4 months after treatment with TAFINLAR in combination with trametinib. Advise patients to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, while taking TAFINLAR [see Drug Interactions (7.2), Use in Specific Populations (8.6)]. 6 ADVERSE REACTIONS The following adverse reactions are discussed in greater detail in another section of the label: • New Primary Malignancies [see Warnings and Precautions (5.1)] • Tumor Promotion in BRAF Wild-Type Melanoma [see Warnings and Precautions (5.2)] • Hemorrhage [see Warnings and Precautions (5.3)] • Venous Thromboembolism [see Warnings and Precautions (5.4)] • Cardiomyopathy [see Warnings and Precautions (5.5)] • Ocular Toxicities [see Warnings and Precautions (5.6)] • Serious Febrile Reactions [see Warnings and Precautions (5.7)] • Serious Skin Toxicity [see Warnings and Precautions (5.8)] • Hyperglycemia [see Warnings and Precautions (5.9)] • Glucose-6-Phosphate Dehydrogenase Deficiency [see Warnings and Precautions (5.10)] 6.1 Clinical Trials Experience Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The data described in the Warnings and Precautions section and below reflect exposure to TAFINLAR as a single agent and in combination withtrametinib. BRAF V600E Unresectable or Metastatic Melanoma: The safety of TAFINLAR as a single agent was evaluated in 586 patients with BRAF V600 mutation-positive unresectable or metastatic melanoma, previously treated or untreated, who received TAFINLAR 150 mg orally twice daily until disease progression or unacceptable toxicity, including 181 patients treated for at least 6 months and 86 additional patients treated for more than 12 months. TAFINLAR was studied in open-label, single-arm trials and in an open-label, randomized, active-controlled trial. The median daily dose of TAFINLAR was 300 mg (range: 118 to 300 mg). Table 3 and Table 4 present adverse drug reactions and laboratory abnormalities identified from analyses of Trial 1 [see Clinical Studies (14.1)]. Trial 1, a multicenter, international, open-label, randomized (3:1), controlled trial allocated 250 patients with unresectable or metastatic BRAF V600E mutation-positive melanoma to receive TAFINLAR 150 mg orally twice daily (n = 187) or dacarbazine 1,000 mg/m2 intravenously every 3 weeks (n = 63). The trial excluded patients with abnormal left ventricular ejection fraction or cardiac valve morphology (≥Grade 2), corrected QT interval ≥480 milliseconds on electrocardiogram, or a known history of glucose-6-phosphate dehydrogenase deficiency. The median duration on treatment was 4.9 months for patients treated with TAFINLAR and 2.8 months for dacarbazine-treated patients. The population exposed to TAFINLAR was 60% male, 99% white, and had a median age of 53 years. The most commonly occurring adverse reactions (≥20%) in patients treated with TAFINLAR were, in order of decreasing frequency: hyperkeratosis, headache, pyrexia, arthralgia, papilloma, alopecia, and palmar-plantar erythrodysesthesia syndrome (PPES). The incidence of adverse events resulting in permanent discontinuation of study medication in Trial 1 was 3% for patients treated with TAFINLAR and 3% for patients treated with dacarbazine. The most frequent (≥2%) adverse reactions leading to dose reduction of TAFINLAR were pyrexia (9%), PPES (3%), chills (3%), fatigue (2%), and headache (2%). Table 3. Selected Common Adverse Reactions Occurring in ≥10% (All Grades) or ≥2% (Grades 3 or 4) of Patients Treated With TAFINLARa
bGrade 4 adverse reactions limited to hyperkeratosis (n = 1) and constipation (n = 1). cIncludes skin papilloma and papilloma. dIncludes squamous cell carcinoma of the skin and keratoacanthoma. eCases of cutaneous squamous cell carcinoma were required to be reported as Grade 3 per protocol. fNA = not applicable. Table 4. Incidence of Laboratory Abnormalities Increased From Baseline Occurring at a Higher Incidence in Patients Treated With TAFINLAR in Trial 1 [Between-Arm Difference of ≥5% (All Grades) or ≥2% (Grades 3 or 4)]
Other clinically important adverse reactions observed in <10% of patients (N = 586) treated with TAFINLAR were: Gastrointestinal Disorders: Pancreatitis. Immune System Disorders: Hypersensitivity manifesting as bullous rash. Renal and Urinary Disorders: Interstitial nephritis. BRAF V600E or V600K Unresectable or Metastatic Melanoma: The safety of TAFINLAR in combination with trametinib was evaluated in Trial 2 and other trials consisting of a total of 202 patients with BRAF V600 mutation-positive unresectable or metastatic melanoma who received TAFINLAR 150 mg orally twice daily in combination with trametinib 2 mg orally once daily until disease progression or unacceptable toxicity. Among these 202 patients, 66 (33%) were exposed to TAFINLAR and 68 (34%) were exposed to trametinib for greater than 6 to 12 months while 40 (20%) were exposed to TAFINLAR and 36 (18%) were exposed to trametinib for greater than one year. The median age was 54 years, 57% were male, and >99% were white. Table 5 presents adverse reactions from Trial 2, a multicenter, open-label, randomized trial of 162 patients with BRAF V600E or V600K mutation-positive melanoma receiving TAFINLAR 150 mg twice daily in combination with trametinib 2 mg orally once daily (n = 55), TAFINLAR 150 mg orally twice daily in combination with trametinib 1 mg once daily (n = 54), and TAFINLAR as a single agent 150 mg orally twice daily (n = 53) [see Clinical Studies (14.2)]. Patients with abnormal LVEF, history of acute coronary syndrome within 6 months, current evidence of Class II or greater congestive heart failure (New York Heart Association), history RVO or RPED, QTc interval ≥480 msec, treatment refractory hypertension, uncontrolled arrhythmias, history of pneumonitis or interstitial lung disease, or a known history of G6PD deficiency were excluded. The median duration of treatment was 10.9 months for both TAFINLAR and trametinib (2-mg orally once-daily treatment group) when used in combination, 10.6 months for both TAFINLAR and trametinib (1-mg orally once-daily treatment group) when used in combination, and 6.1 months for TAFINLAR as a single agent. In Trial 2, 13% of patients receiving TAFINLAR in combination with trametinib experienced adverse reactions resulting in permanent discontinuation of trial medication(s). The most common adverse reaction resulting in permanent discontinuation was pyrexia (4%). Adverse reactions led to dose reductions in 49% and dose interruptions in 67% of patients treated with TAFINLAR in combination with trametinib. Pyrexia, chills, and nausea were the most common reasons cited for dose reductions and pyrexia, chills, and decreased ejection fraction were the most common reasons cited for dose interruptions of TAFINLAR and trametinib when used in combination. Table 5. Common Adverse Drug Reactions Occurring in ≥10% at (All Grades) or ≥5% (Grades 3 or 4) of Patients Treated With TAFINLAR in Combination With Trametinib in Trial 2
bIncludes the following terms: peripheral edema, edema, and lymphedema. cIncludes the following terms: rash, rash generalized, rash pruritic, rash erythematous, rash papular, rash vesicular, rash macular, and rash maculo-papular. dIncludes the following terms: abdominal pain, abdominal pain upper, abdominal pain lower, and abdominal discomfort. eIncludes the following terms: brain stem hemorrhage, cerebral hemorrhage, gastric hemorrhage, epistaxis, gingival hemorrhage, hematuria, vaginal hemorrhage, hemorrhage intracranial, eye hemorrhage, and vitreous hemorrhage. fIncludes the following terms: renal failure and renal failure acute. Other clinically important adverse reactions (N = 202) observed in <10% of patients treated with TAFINLAR in combination with trametinib were Eye Disorders: Vision blurred, transient blindness. Gastrointestinal Disorders: Stomatitis, pancreatitis. General Disorders and Administration Site Conditions: Asthenia. Infections and Infestations: Cellulitis, folliculitis, paronychia, rash pustular. Neoplasms Benign, Malignant, and Unspecified (including cysts and polyps): Skin papilloma. Skin and Subcutaneous Tissue Disorders: Palmar-plantar erythrodysesthesia syndrome, hyperkeratosis, hyperhidrosis. Vascular Disorders: Hypertension. Table 6. Treatment-Emergent Laboratory Abnormalities Occurring at ≥10% (All Grades) or ≥2% (Grades 3 or 4)] of Patients Treated With TAFINLAR in Combination With Trametinib in Trial 2
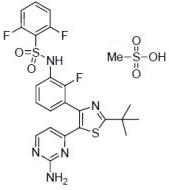
ALT = Alanine aminotransferase; AST = Aspartate aminotransferase; GGT = Gamma glutamyltransferase. QT Prolongation: In Trial 2, QTcF prolongation to >500 msec occurred in 4% (2/55) of patients treated with TAFINLAR in combination with trametinib and in 2% (1/53) of patients treated with TAFINLAR as a single agent. The QTcF was increased more than 60 msec from baseline in 13% (7/55) of patients treated with TAFINLAR in combination with trametinib and 2% (1/53) of patients treated with TAFINLAR as a single agent. 7 DRUG INTERACTIONS 7.1 Effects of Other Drugs on Dabrafenib Dabrafenib is primarily metabolized by CYP2C8 and CYP3A4. Strong inhibitors of CYP3A4 or CYP2C8 may increase concentrations of dabrafenib and strong inducers of CYP3A4 or CYP2C8 may decrease concentrations of dabrafenib [see Clinical Pharmacology (12.3)]. Substitution of strong inhibitors or strong inducers of CYP3A4 or CYP2C8 is recommended during treatment with TAFINLAR. If concomitant use of strong inhibitors (e.g., ketoconazole, nefazodone, clarithromycin, gemfibrozil) or strong inducers (e.g., rifampin, phenytoin, carbamazepine, phenobarbital, St John’s wort) of CYP3A4 or CYP2C8 is unavoidable, monitor patients closely for adverse reactions when taking strong inhibitors or loss of efficacy when taking strong inducers. 7.2 Effects of Dabrafenib on Other Drugs Dabrafenib induces CYP3A4 and CYP2C9. Dabrafenib decreased the systemic exposures of midazolam (a CYP3A4 substrate), S-warfarin (a CYP2C9 substrate), and R-warfarin (a CYP3A4/CYP1A2 substrate) [see Clinical Pharmacology (12.3)]. Monitor international normalized ratio (INR) levels more frequently in patients receiving warfarin during initiation or discontinuation of dabrafenib. Coadministration of TAFINLAR with other substrates of these enzymes, including dexamethasone or hormonal contraceptives, can result in decreased concentrations and loss of efficacy [see Use in Specific Populations (8.1, 8.6)]. Substitute for these medications or monitor patients for loss of efficacy if use of these medications is unavoidable. 7.3 Trametinib Coadministration of TAFINLAR 150 mg twice daily and trametinib 2 mg once daily resulted in no clinically relevant pharmacokinetic drug interactions [see Clinical Pharmacology (12.3)]. 8 USE IN SPECIFIC POPULATIONS 8.1 Pregnancy Pregnancy Category D Risk Summary: Based on its mechanism of action, TAFINLAR can cause fetal harm when administered to a pregnant woman. Dabrafenib was teratogenic and embryotoxic in rats at doses three times greater than the human exposure at the recommended clinical dose of 150 mg twice daily based on AUC. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Warnings and Precautions (5.11)]. Animal Data: In a combined female fertility and embryofetal development study in rats, developmental toxicity consisted of embryo-lethality, ventricular septal defects, and variation in thymic shape at a dabrafenib dose of 300 mg/kg/day (approximately three times the human exposure at the recommended dose based on AUC). At doses of 20 mg/kg/day or greater (equivalent to the human exposure at the recommended dose based on AUC), rats demonstrated delays in skeletal development and reduced fetal body weight. 8.3 Nursing Mothers It is not known whether this drug is present in human milk. Because many drugs are present in human milk and because of the potential for serious adverse reactions from TAFINLAR in nursing infants, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother. 8.4 Pediatric Use The safety and effectiveness of TAFINLAR have not been established in pediatric patients. In a repeat-dose toxicity study in juvenile rats, an increased incidence of kidney cysts and tubular deposits were noted at doses as low as 0.2 times the human exposure at the recommended adult dose based on AUC. Additionally, forestomach hyperplasia, decreased bone length and early vaginal opening were noted at doses as low as 0.8 times the human exposure at the recommended adult dose based on AUC. 8.5 Geriatric Use One hundred and twenty-six (22%) of 586 patients in clinical trials of TAFINLAR administered as a single agent and 40 (21%) of the 187 patients receiving TAFINLAR in Trial 1 were ≥65 years of age. No overall differences in the effectiveness or safety of TAFINLAR were observed in the elderly in Trial 1. Across all clinical trials of TAFINLAR administered in combination with trametinib, there was an insufficient number of patients aged 65 years and over to determine whether they respond differently from younger patients. In Trial 2, 11 patients (20%) were 65 years of age and older, and 2 patients (4%) were 75 years of age and older. 8.6 Females and Males of Reproductive Potential Contraception:Females: Advise female patients of reproductive potential to use highly effective contraception during treatment and for at least 2 weeks after the last dose of TAFINLAR or at least 4 months after the last dose of TAFINLAR taken in combination with trametinib. Counsel patients to use a non-hormonal method of contraception since TAFINLAR can render hormonal contraceptives ineffective. Advise patients to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, while taking TAFINLAR [see Warnings and Precautions (5.11), Drug Interactions (7.1), Use in Specific Populations (8.1)]. Infertility: Females: Increased follicular cysts and decreased corpora lutea were observed in female rats treated with trametinib. Advise female patients of reproductive potential that TAFINLAR taken in combination with trametinib may impair fertility in female patients. Males: Effects on spermatogenesis have been observed in animals. Advise male patients of the potential risk for impaired spermatogenesis, and to seek counseling on fertility and family planning options prior to starting treatment with TAFINLAR [see Nonclinical Toxicology (13.1)]. 8.7 Hepatic Impairment No formal pharmacokinetic trial in patients with hepatic impairment has been conducted. Dose adjustment is not recommended for patients with mild hepatic impairment based on the results of the population pharmacokinetic analysis. As hepatic metabolism and biliary secretion are the primary routes of elimination of dabrafenib and its metabolites, patients with moderate to severe hepatic impairment may have increased exposure. An appropriate dose has not been established for patients with moderate to severe hepatic impairment [see Clinical Pharmacology (12.3)]. 8.8 Renal Impairment No formal pharmacokinetic trial in patients with renal impairment has been conducted. Dose adjustment is not recommended for patients with mild or moderate renal impairment based on the results of the population pharmacokinetic analysis. An appropriate dose has not been established for patients with severe renal impairment [see Clinical Pharmacology (12.3)]. 10 OVERDOSAGE There is no information on overdosage of TAFINLAR. Since dabrafenib is highly bound to plasma proteins, hemodialysis is likely to be ineffective in the treatment of overdose with TAFINLAR. 11 DESCRIPTION Dabrafenib mesylate is a kinase inhibitor. The chemical name for dabrafenib mesylate is N-{3-[5-(2-Amino-4-pyrimidinyl)-2-(1,1-dimethylethyl)-1,3-thiazol-4-yl]-2-fluorophenyl}-2,6-difluorobenzene sulfonamide, methanesulfonate salt. It has the molecular formula C23H20F3N5O2S2•CH4O3S and a molecular weight of 615.68. Dabrafenib mesylate has the following chemical structure.
Dabrafenib mesylate is a white to slightly colored solid with three pKas:
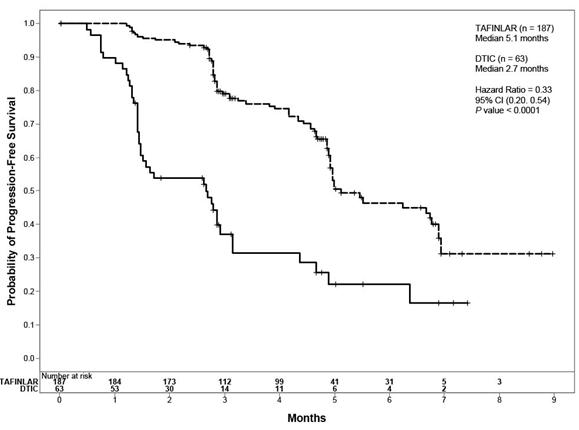
bStratified log-rank test. CI = Confidence interval; CR = Complete response; HR = Hazard ratio; NR = Not reached; PR = Partial response. Figure 1. Kaplan-Meier Curves of Investigator-Assessed Progression-Free Survival
14.2 BRAF V600E or V600K Unresectable or Metastatic Melanoma Trial 2 was a multicenter, open-label, randomized (1:1:1) dose-ranging trial designed to evaluate the clinical activity and safety of TAFINLAR in combination with trametinib (at two different doses) and to compare the safety with TAFINLAR as a single agent in 162 patients with BRAF V600E or V600K mutation-positive, unresectable or metastatic melanoma. Patients were permitted to have had one prior chemotherapy regimen and prior aldesleukin; patients with prior exposure to BRAF or MEK inhibitors were ineligible. Patients were randomized to receive TAFINLAR 150 mg orally twice daily with trametinib 2 mg orally once daily (n = 54), TAFINLAR 150 mg orally twice daily with trametinib 1 mg orally once daily (n = 54), or TAFINLAR 150 mg orally twice daily (n = 54). Treatment continued until disease progression or unacceptable toxicity. Patients randomized to TAFINLAR as a single agent were offered TAFINLAR 150 mg orally twice daily with trametinib 2 mg orally once daily at the time of investigator-assessed disease progression. The major efficacy outcome measure was investigator-assessed overall response rate (ORR). Additional efficacy outcome measures were investigator-assessed duration of response, independent radiology review committee (IRRC)-assessed ORR, and IRRC-assessed duration of response. The median age of patients was 53 years, 57% were male, >99% were white, 66% of patients had a pre-treatment ECOG performance status of 0, 67% had M1c disease, 54% had a normal LDH at baseline, and 8% had history of brain metastases. Most patients (81%) had not received prior anti-cancer therapy for unresectable or metastatic disease. Based on local laboratory or centralized testing, 85% of patients’ tumors had BRAF V600E mutations and 15% had BRAF V600K mutations. The median duration of follow-up was 14 months. Efficacy outcomes for the trial arms receiving TAFINLAR in combination with trametinib 2 mg orally once daily and TAFINLAR as a single agent are summarized in Table 9. Table 9. Investigator-Assessed and Independent Review Committee-Assessed Response Rates and Response Duration in Trial 2
The ORR results were similar in subgroups defined by BRAF mutation subtype, i.e., in the 85% of patients with V600E mutation-positive melanoma and in the 15% of patients with V600K mutation-positive melanoma. In exploratory subgroup analyses of the patients with retrospectively confirmed BRAF V600E or V600K mutation-positive melanoma using the THxID™-BRAF assay, the ORR results were also similar to the intent-to-treat analysis. 16 HOW SUPPLIED/STORAGE AND HANDLING 50 mg Capsules: Dark red capsule imprinted with ‘GS TEW’ and ‘50 mg’ available in bottles of 120 (NDC 0173-0846-08). Each bottle contains a silica gel desiccant. 75 mg Capsules: Dark pink capsule imprinted with ‘GS LHF’ and ‘75 mg’ available in bottles of 120 (NDC 0173-0847-08). Each bottle contains a silica gel desiccant. Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. 17 PATIENT COUNSELING INFORMATION See FDA-approved patient labeling (Medication Guide). Inform patients of the following: • Evidence of BRAF V600E mutation in the tumor specimen is necessary to identify patients for whom treatment with TAFINLAR as a single agent is indicated and evidence of BRAF V600E or V600K mutation in tumor specimens is necessary to identify patients for whom treatment with TAFINLAR in combination with trametinib is indicated [see Dosage and Administration (2.1)]. • TAFINLAR increases the risk of developing new primary cutaneous and non-cutaneous malignancies. Advise patients to contact their doctor immediately for any new lesions, changes to existing lesions on their skin, or signs and symptoms of other malignancies [see Warnings and Precautions (5.1)]. • TAFINLAR administered in combination with trametinib increases the risk of intracranial and gastrointestinal hemorrhage. Advise patients to contact their healthcare provider to seek immediate medical attention for signs or symptoms of unusual bleeding or hemorrhage [see Warnings and Precautions (5.3)]. • TAFINLAR administered in combination with trametinib increases the risks of pulmonary embolism and deep venous thrombosis. Advise patients to seek immediate medical attention for sudden onset of difficulty breathing, leg pain, or swelling [see Warnings and Precautions (5.4)]. • TAFINLAR administered in combination with trametinib can cause cardiomyopathy. Advise patients to immediately report any signs or symptoms of heart failure to their healthcare provider [see Warnings and Precautions (5.5)]. • TAFINLAR can cause visual disturbances; TAFINLAR administered in combination with trametinib can lead to blindness. Advise patients to contact their healthcare provider if they experience any changes in their vision [see Warnings and Precautions (5.6)]. • TAFINLAR administered as a single agent and in combination with trametinib, can cause pyrexia including serious febrile reactions. Inform patients that the incidence and severity of pyrexia are increased when TAFINLAR is given in combination with trametinib. Instruct patients to contact their doctor if they develop fever while taking TAFINLAR [see Warnings and Precautions (5.7)]. • TAFINLAR in combination with trametinib can cause serious skin toxicities which may require hospitalization. Advise patients to contact their healthcare provider for progressive or intolerable rash [see Warnings and Precautions (5.8) ]. • TAFINLAR can impair glucose control in diabetic patients resulting in the need for more intensive hypoglycemic treatment. Advise patients to contact their doctor to report symptoms of severe hyperglycemia [see Warnings and Precautions (5.9)]. • TAFINLAR may cause hemolytic anemia in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Advise patients with known G6PD deficiency to contact their doctor to report signs or symptoms of anemia or hemolysis [see Warnings and Precautions (5.10)]. • TAFINLAR can cause fetal harm if taken during pregnancy. Instruct female patients to use non-hormonal, highly effective contraception during treatment and for 2 weeks after discontinuation of treatment with TAFINLAR as a single agent, or for 4 months after discontinuation of treatment with TAFINLAR in combination with trametinib. Advise patients to contact their doctor if they become pregnant, or if pregnancy is suspected, while taking TAFINLAR [see Warnings and Precautions (5.11), Use in Specific Populations (8.1)]. • Nursing infants may experience serious adverse reactions if the mother is taking TAFINLAR during breastfeeding. Advise breastfeeding mothers to discontinue nursing while taking TAFINLAR [see Use in Specific Populations (8.3)]. • Male patients are at an increased risk for impaired spermatogenesis [see Use in Specific Populations (8.6)]. • TAFINLAR should be taken either at least 1 hour before or at least 2 hours after a meal [see Dosage and Administration (2.1)]. TAFINLAR is a registered trademark of the GlaxoSmithKline group of companies. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||